 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTree-of-Evidence: Efficient "System 2" Search for Faithful Multimodal Grounding
Apr 09, 2026Large Multimodal Models (LMMs) achieve state-of-the-art performance in high-stakes domains like healthcare, yet their reasoning remains opaque. Current interpretability methods, such as attention mechanisms or post-hoc saliency, often fail to faithfully represent the model's decision-making process, particularly when integrating heterogeneous modalities like time-series and text. We introduce Tree-of-Evidence (ToE), an inference-time search algorithm that frames interpretability as a discrete optimization problem. Rather than relying on soft attention weights, ToE employs lightweight Evidence Bottlenecks that score coarse groups or units of data (e.g., vital-sign windows, report sentences) and performs a beam search to identify the compact evidence set required to reproduce the model's prediction. We evaluate ToE across six tasks spanning three datasets and two domains: four clinical prediction tasks on MIMIC-IV, cross-center validation on eICU, and non-clinical fault detection on LEMMA-RCA. ToE produces auditable evidence traces while maintaining predictive performance, retaining over 0.98 of full-model AUROC with as few as five evidence units across all settings. Under sparse evidence budgets, ToE achieves higher decision agreement and lower probability fidelity error than other approaches. Qualitative analyses show that ToE adapts its search strategy: it often resolves straightforward cases using only vitals, while selectively incorporating text when physiological signals are ambiguous. ToE therefore provides a practical mechanism for auditing multimodal models by revealing which discrete evidence units support each prediction.
EvidenceRL: Reinforcing Evidence Consistency for Trustworthy Language Models
Mar 20, 2026Large Language Models (LLMs) are fluent but prone to hallucinations, producing answers that appear plausible yet are unsupported by available evidence. This failure is especially problematic in high-stakes domains where decisions must be justified by verifiable information. We introduce \textbf{EvidenceRL}, a reinforcement learning framework that enforces evidence adherence during training. EvidenceRL scores candidate responses for grounding (entailment with retrieved evidence and context) and correctness (agreement with reference answers) and optimizes the generator using Group Relative Policy Optimization (GRPO). We evaluate across two high-stakes domains, cardiac diagnosis and legal reasoning, where EvidenceRL consistently improves evidence grounding and faithfulness without sacrificing task accuracy. On cardiac diagnosis, F1@3 increases from 37.0 to 54.5 on Llama-3.2-3B while grounding ($G_{\max}@3$) rises from 47.6 to 78.2; hallucinations drop nearly 5$\times$ and evidence-supported diagnoses increase from 31.8\% to 61.6\%. On legal reasoning, EvidenceRL raises Faithfulness from 32.8\% to 67.6\% on Llama-3.1-8B, demonstrating consistent behavioral change across domains. Our code is open-sourced at https://github.com/Wizaaard/EvidenceRL.git.
Monocular Markerless Motion Capture Enables Quantitative Assessment of Upper Extremity Reachable Workspace
Feb 13, 2026To validate a clinically accessible approach for quantifying the Upper Extremity Reachable Workspace (UERW) using a single (monocular) camera and Artificial Intelligence (AI)-driven Markerless Motion Capture (MMC) for biomechanical analysis. Objective assessment and validation of these techniques for specific clinically oriented tasks are crucial for their adoption in clinical motion analysis. AI-driven monocular MMC reduces the barriers to adoption in the clinic and has the potential to reduce the overhead for analysis of this common clinical assessment. Nine adult participants with no impairments performed the standardized UERW task, which entails reaching targets distributed across a virtual sphere centered on the torso, with targets displayed in a VR headset. Movements were simultaneously captured using a marker-based motion capture system and a set of eight FLIR cameras. We performed monocular video analysis on two of these video camera views to compare a frontal and offset camera configurations. The frontal camera orientation demonstrated strong agreement with the marker-based reference, exhibiting a minimal mean bias of $0.61 \pm 0.12$ \% reachspace reached per octanct (mean $\pm$ standard deviation). In contrast, the offset camera view underestimated the percent workspace reached ($-5.66 \pm 0.45$ \% reachspace reached). Conclusion: The findings support the feasibility of a frontal monocular camera configuration for UERW assessment, particularly for anterior workspace evaluation where agreement with marker-based motion capture was highest. The overall performance demonstrates clinical potential for practical, single-camera assessments. This study provides the first validation of monocular MMC system for the assessment of the UERW task. By reducing technical complexity, this approach enables broader implementation of quantitative upper extremity mobility assessment.
LLM-as-RNN: A Recurrent Language Model for Memory Updates and Sequence Prediction
Jan 19, 2026Large language models are strong sequence predictors, yet standard inference relies on immutable context histories. After making an error at generation step t, the model lacks an updatable memory mechanism that improves predictions for step t+1. We propose LLM-as-RNN, an inference-only framework that turns a frozen LLM into a recurrent predictor by representing its hidden state as natural-language memory. This state, implemented as a structured system-prompt summary, is updated at each timestep via feedback-driven text rewrites, enabling learning without parameter updates. Under a fixed token budget, LLM-as-RNN corrects errors and retains task-relevant patterns, effectively performing online learning through language. We evaluate the method on three sequential benchmarks in healthcare, meteorology, and finance across Llama, Gemma, and GPT model families. LLM-as-RNN significantly outperforms zero-shot, full-history, and MemPrompt baselines, improving predictive accuracy by 6.5% on average, while producing interpretable, human-readable learning traces absent in standard context accumulation.
MedAgentGym: Training LLM Agents for Code-Based Medical Reasoning at Scale
Jun 04, 2025



We introduce MedAgentGYM, the first publicly available training environment designed to enhance coding-based medical reasoning capabilities in large language model (LLM) agents. MedAgentGYM comprises 72,413 task instances across 129 categories derived from authentic real-world biomedical scenarios. Tasks are encapsulated within executable coding environments, each featuring detailed task descriptions, interactive feedback mechanisms, verifiable ground-truth annotations, and scalable training trajectory generation. Extensive benchmarking of over 30 LLMs reveals a notable performance disparity between commercial API-based models and open-source counterparts. Leveraging MedAgentGYM, Med-Copilot-7B achieves substantial performance gains through supervised fine-tuning (+36.44%) and continued reinforcement learning (+42.47%), emerging as an affordable and privacy-preserving alternative competitive with gpt-4o. By offering both a comprehensive benchmark and accessible, expandable training resources within unified execution environments, MedAgentGYM delivers an integrated platform to develop LLM-based coding assistants for advanced biomedical research and practice.
Novel Extraction of Discriminative Fine-Grained Feature to Improve Retinal Vessel Segmentation
May 06, 2025Retinal vessel segmentation is a vital early detection method for several severe ocular diseases. Despite significant progress in retinal vessel segmentation with the advancement of Neural Networks, there are still challenges to overcome. Specifically, retinal vessel segmentation aims to predict the class label for every pixel within a fundus image, with a primary focus on intra-image discrimination, making it vital for models to extract more discriminative features. Nevertheless, existing methods primarily focus on minimizing the difference between the output from the decoder and the label, but ignore fully using feature-level fine-grained representations from the encoder. To address these issues, we propose a novel Attention U-shaped Kolmogorov-Arnold Network named AttUKAN along with a novel Label-guided Pixel-wise Contrastive Loss for retinal vessel segmentation. Specifically, we implement Attention Gates into Kolmogorov-Arnold Networks to enhance model sensitivity by suppressing irrelevant feature activations and model interpretability by non-linear modeling of KAN blocks. Additionally, we also design a novel Label-guided Pixel-wise Contrastive Loss to supervise our proposed AttUKAN to extract more discriminative features by distinguishing between foreground vessel-pixel pairs and background pairs. Experiments are conducted across four public datasets including DRIVE, STARE, CHASE_DB1, HRF and our private dataset. AttUKAN achieves F1 scores of 82.50%, 81.14%, 81.34%, 80.21% and 80.09%, along with MIoU scores of 70.24%, 68.64%, 68.59%, 67.21% and 66.94% in the above datasets, which are the highest compared to 11 networks for retinal vessel segmentation. Quantitative and qualitative results show that our AttUKAN achieves state-of-the-art performance and outperforms existing retinal vessel segmentation methods. Our code will be available at https://github.com/stevezs315/AttUKAN.
Advancing Problem-Based Learning in Biomedical Engineering in the Era of Generative AI
Mar 20, 2025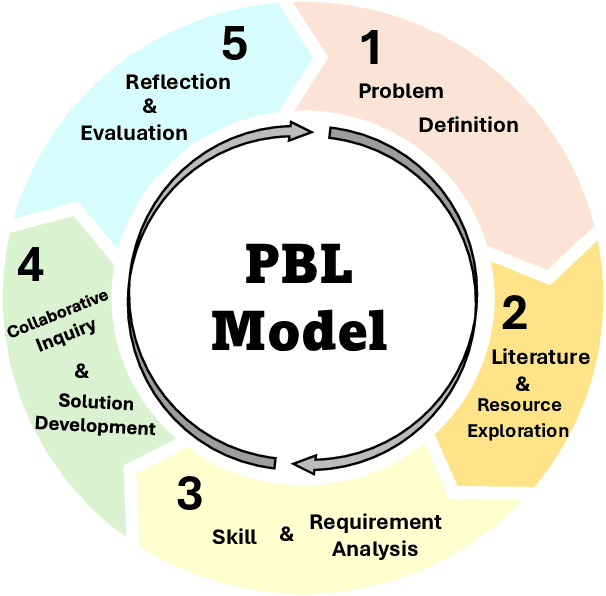

Problem-Based Learning (PBL) has significantly impacted biomedical engineering (BME) education since its introduction in the early 2000s, effectively enhancing critical thinking and real-world knowledge application among students. With biomedical engineering rapidly converging with artificial intelligence (AI), integrating effective AI education into established curricula has become challenging yet increasingly necessary. Recent advancements, including AI's recognition by the 2024 Nobel Prize, have highlighted the importance of training students comprehensively in biomedical AI. However, effective biomedical AI education faces substantial obstacles, such as diverse student backgrounds, limited personalized mentoring, constrained computational resources, and difficulties in safely scaling hands-on practical experiments due to privacy and ethical concerns associated with biomedical data. To overcome these issues, we conducted a three-year (2021-2023) case study implementing an advanced PBL framework tailored specifically for biomedical AI education, involving 92 undergraduate and 156 graduate students from the joint Biomedical Engineering program of Georgia Institute of Technology and Emory University. Our approach emphasizes collaborative, interdisciplinary problem-solving through authentic biomedical AI challenges. The implementation led to measurable improvements in learning outcomes, evidenced by high research productivity (16 student-authored publications), consistently positive peer evaluations, and successful development of innovative computational methods addressing real biomedical challenges. Additionally, we examined the role of generative AI both as a teaching subject and an educational support tool within the PBL framework. Our study presents a practical and scalable roadmap for biomedical engineering departments aiming to integrate robust AI education into their curricula.
MedAdapter: Efficient Test-Time Adaptation of Large Language Models towards Medical Reasoning
May 05, 2024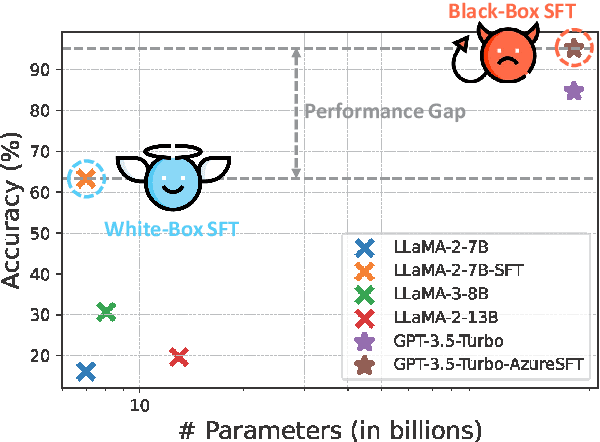
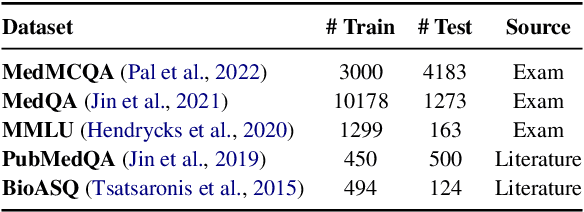
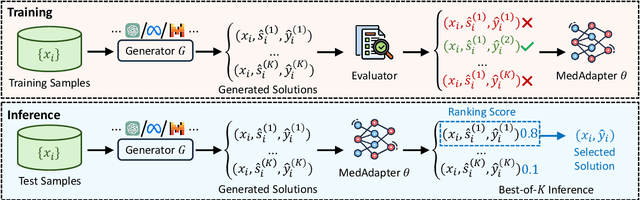
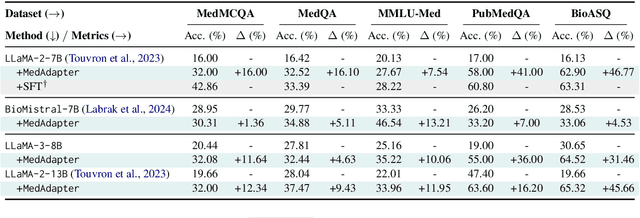
Despite their improved capabilities in generation and reasoning, adapting large language models (LLMs) to the biomedical domain remains challenging due to their immense size and corporate privacy. In this work, we propose MedAdapter, a unified post-hoc adapter for test-time adaptation of LLMs towards biomedical applications. Instead of fine-tuning the entire LLM, MedAdapter effectively adapts the original model by fine-tuning only a small BERT-sized adapter to rank candidate solutions generated by LLMs. Experiments demonstrate that MedAdapter effectively adapts both white-box and black-box LLMs in biomedical reasoning, achieving average performance improvements of 25.48% and 11.31%, respectively, without requiring extensive computational resources or sharing data with third parties. MedAdapter also yields superior performance when combined with train-time adaptation, highlighting a flexible and complementary solution to existing adaptation methods. Faced with the challenges of balancing model performance, computational resources, and data privacy, MedAdapter provides an efficient, privacy-preserving, cost-effective, and transparent solution for adapting LLMs to the biomedical domain.
BMRetriever: Tuning Large Language Models as Better Biomedical Text Retrievers
Apr 29, 2024



Developing effective biomedical retrieval models is important for excelling at knowledge-intensive biomedical tasks but still challenging due to the deficiency of sufficient publicly annotated biomedical data and computational resources. We present BMRetriever, a series of dense retrievers for enhancing biomedical retrieval via unsupervised pre-training on large biomedical corpora, followed by instruction fine-tuning on a combination of labeled datasets and synthetic pairs. Experiments on 5 biomedical tasks across 11 datasets verify BMRetriever's efficacy on various biomedical applications. BMRetriever also exhibits strong parameter efficiency, with the 410M variant outperforming baselines up to 11.7 times larger, and the 2B variant matching the performance of models with over 5B parameters. The training data and model checkpoints are released at \url{https://huggingface.co/BMRetriever} to ensure transparency, reproducibility, and application to new domains.
RAM-EHR: Retrieval Augmentation Meets Clinical Predictions on Electronic Health Records
Feb 25, 2024



We present RAM-EHR, a Retrieval AugMentation pipeline to improve clinical predictions on Electronic Health Records (EHRs). RAM-EHR first collects multiple knowledge sources, converts them into text format, and uses dense retrieval to obtain information related to medical concepts. This strategy addresses the difficulties associated with complex names for the concepts. RAM-EHR then augments the local EHR predictive model co-trained with consistency regularization to capture complementary information from patient visits and summarized knowledge. Experiments on two EHR datasets show the efficacy of RAM-EHR over previous knowledge-enhanced baselines (3.4% gain in AUROC and 7.2% gain in AUPR), emphasizing the effectiveness of the summarized knowledge from RAM-EHR for clinical prediction tasks. The code will be published at \url{https://github.com/ritaranx/RAM-EHR}.