 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Policy to Logic for Efficient and Interpretable Coverage Assessment
Jan 08, 2026Large Language Models (LLMs) have demonstrated strong capabilities in interpreting lengthy, complex legal and policy language. However, their reliability can be undermined by hallucinations and inconsistencies, particularly when analyzing subjective and nuanced documents. These challenges are especially critical in medical coverage policy review, where human experts must be able to rely on accurate information. In this paper, we present an approach designed to support human reviewers by making policy interpretation more efficient and interpretable. We introduce a methodology that pairs a coverage-aware retriever with symbolic rule-based reasoning to surface relevant policy language, organize it into explicit facts and rules, and generate auditable rationales. This hybrid system minimizes the number of LLM inferences required which reduces overall model cost. Notably, our approach achieves a 44% reduction in inference cost alongside a 4.5% improvement in F1 score, demonstrating both efficiency and effectiveness.
Zero-shot Medical Event Prediction Using a Generative Pre-trained Transformer on Electronic Health Records
Mar 07, 2025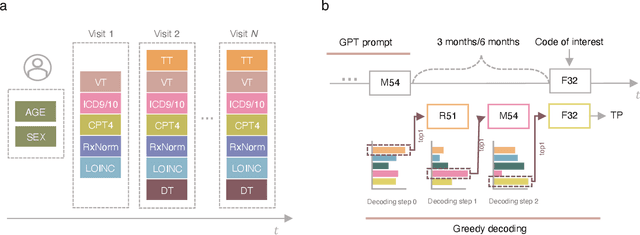
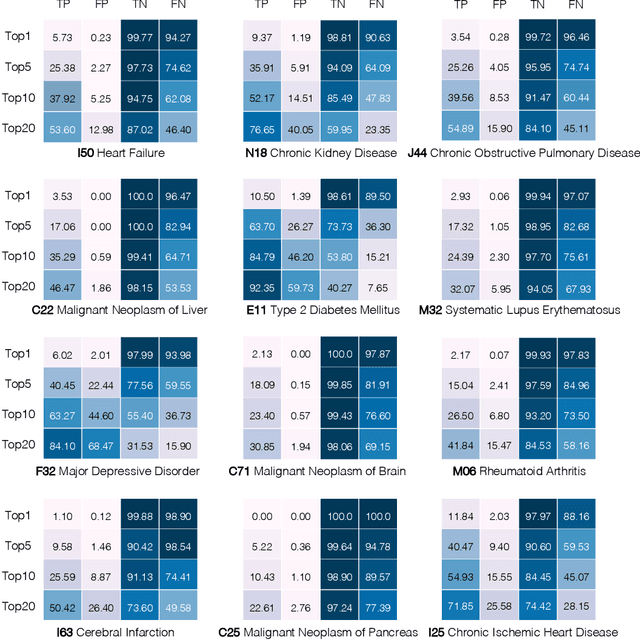
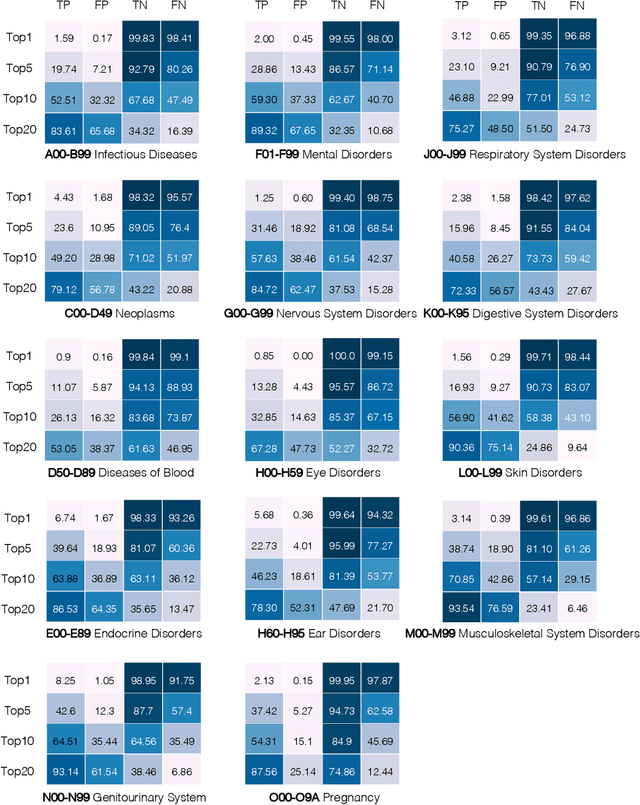
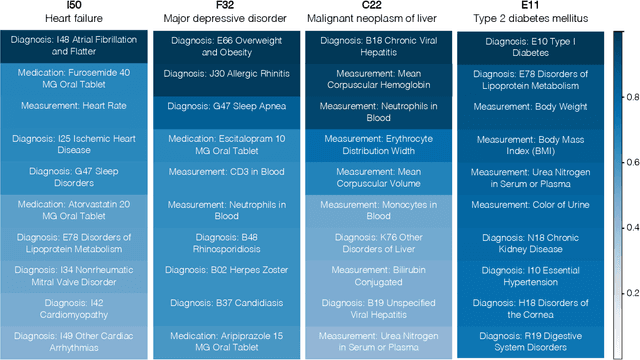
Longitudinal data in electronic health records (EHRs) represent an individual`s clinical history through a sequence of codified concepts, including diagnoses, procedures, medications, and laboratory tests. Foundational models, such as generative pre-trained transformers (GPT), can leverage this data to predict future events. While fine-tuning of these models enhances task-specific performance, it is costly, complex, and unsustainable for every target. We show that a foundation model trained on EHRs can perform predictive tasks in a zero-shot manner, eliminating the need for fine-tuning. This study presents the first comprehensive analysis of zero-shot forecasting with GPT-based foundational models in EHRs, introducing a novel pipeline that formulates medical concept prediction as a generative modeling task. Unlike supervised approaches requiring extensive labeled data, our method enables the model to forecast a next medical event purely from a pretraining knowledge. We evaluate performance across multiple time horizons and clinical categories, demonstrating model`s ability to capture latent temporal dependencies and complex patient trajectories without task supervision. Model performance for predicting the next medical concept was evaluated using precision and recall metrics, achieving an average top1 precision of 0.614 and recall of 0.524. For 12 major diagnostic conditions, the model demonstrated strong zero-shot performance, achieving high true positive rates while maintaining low false positives. We demonstrate the power of a foundational EHR GPT model in capturing diverse phenotypes and enabling robust, zero-shot forecasting of clinical outcomes. This capability enhances the versatility of predictive healthcare models and reduces the need for task-specific training, enabling more scalable applications in clinical settings.
Cross-Modality Translation with Generative Adversarial Networks to Unveil Alzheimer's Disease Biomarkers
May 08, 2024Generative approaches for cross-modality transformation have recently gained significant attention in neuroimaging. While most previous work has focused on case-control data, the application of generative models to disorder-specific datasets and their ability to preserve diagnostic patterns remain relatively unexplored. Hence, in this study, we investigated the use of a generative adversarial network (GAN) in the context of Alzheimer's disease (AD) to generate functional network connectivity (FNC) and T1-weighted structural magnetic resonance imaging data from each other. We employed a cycle-GAN to synthesize data in an unpaired data transition and enhanced the transition by integrating weak supervision in cases where paired data were available. Our findings revealed that our model could offer remarkable capability, achieving a structural similarity index measure (SSIM) of $0.89 \pm 0.003$ for T1s and a correlation of $0.71 \pm 0.004$ for FNCs. Moreover, our qualitative analysis revealed similar patterns between generated and actual data when comparing AD to cognitively normal (CN) individuals. In particular, we observed significantly increased functional connectivity in cerebellar-sensory motor and cerebellar-visual networks and reduced connectivity in cerebellar-subcortical, auditory-sensory motor, sensory motor-visual, and cerebellar-cognitive control networks. Additionally, the T1 images generated by our model showed a similar pattern of atrophy in the hippocampal and other temporal regions of Alzheimer's patients.
Leveraging Pretrained Models for Automatic Summarization of Doctor-Patient Conversations
Sep 24, 2021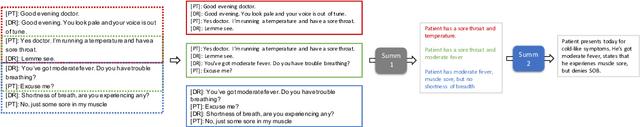

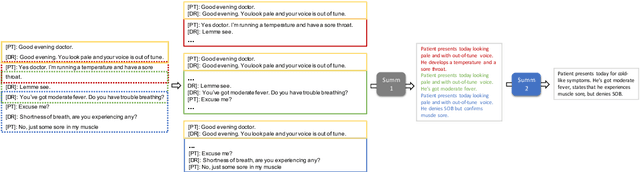
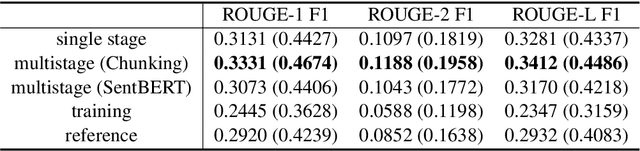
Fine-tuning pretrained models for automatically summarizing doctor-patient conversation transcripts presents many challenges: limited training data, significant domain shift, long and noisy transcripts, and high target summary variability. In this paper, we explore the feasibility of using pretrained transformer models for automatically summarizing doctor-patient conversations directly from transcripts. We show that fluent and adequate summaries can be generated with limited training data by fine-tuning BART on a specially constructed dataset. The resulting models greatly surpass the performance of an average human annotator and the quality of previous published work for the task. We evaluate multiple methods for handling long conversations, comparing them to the obvious baseline of truncating the conversation to fit the pretrained model length limit. We introduce a multistage approach that tackles the task by learning two fine-tuned models: one for summarizing conversation chunks into partial summaries, followed by one for rewriting the collection of partial summaries into a complete summary. Using a carefully chosen fine-tuning dataset, this method is shown to be effective at handling longer conversations, improving the quality of generated summaries. We conduct both an automatic evaluation (through ROUGE and two concept-based metrics focusing on medical findings) and a human evaluation (through qualitative examples from literature, assessing hallucination, generalization, fluency, and general quality of the generated summaries).
DeepDeath: Learning to Predict the Underlying Cause of Death with Big Data
May 06, 2017

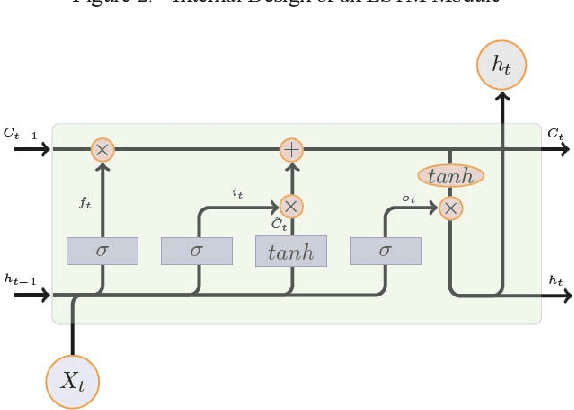
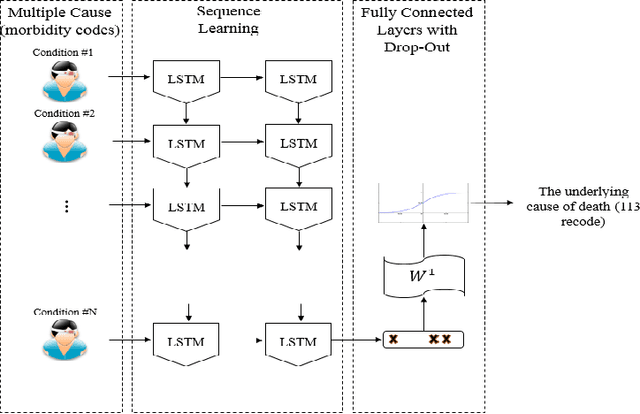
Multiple cause-of-death data provides a valuable source of information that can be used to enhance health standards by predicting health related trajectories in societies with large populations. These data are often available in large quantities across U.S. states and require Big Data techniques to uncover complex hidden patterns. We design two different classes of models suitable for large-scale analysis of mortality data, a Hadoop-based ensemble of random forests trained over N-grams, and the DeepDeath, a deep classifier based on the recurrent neural network (RNN). We apply both classes to the mortality data provided by the National Center for Health Statistics and show that while both perform significantly better than the random classifier, the deep model that utilizes long short-term memory networks (LSTMs), surpasses the N-gram based models and is capable of learning the temporal aspect of the data without a need for building ad-hoc, expert-driven features.
MotifMark: Finding Regulatory Motifs in DNA Sequences
May 04, 2017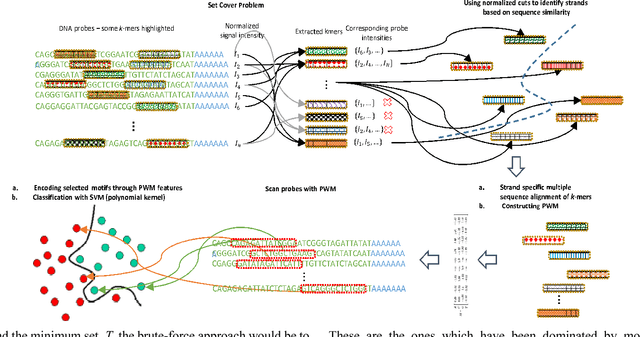
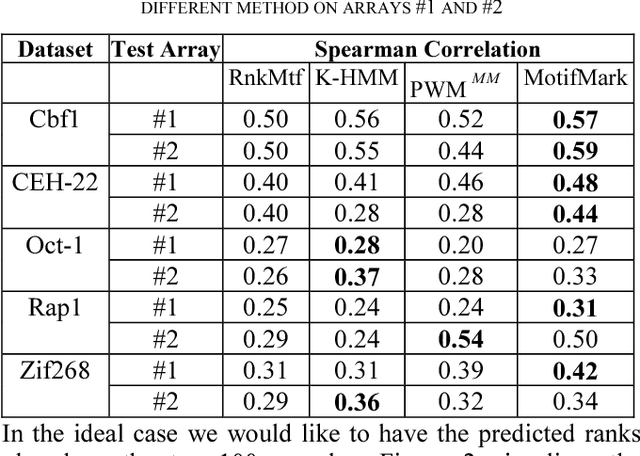

The interaction between proteins and DNA is a key driving force in a significant number of biological processes such as transcriptional regulation, repair, recombination, splicing, and DNA modification. The identification of DNA-binding sites and the specificity of target proteins in binding to these regions are two important steps in understanding the mechanisms of these biological activities. A number of high-throughput technologies have recently emerged that try to quantify the affinity between proteins and DNA motifs. Despite their success, these technologies have their own limitations and fall short in precise characterization of motifs, and as a result, require further downstream analysis to extract useful and interpretable information from a haystack of noisy and inaccurate data. Here we propose MotifMark, a new algorithm based on graph theory and machine learning, that can find binding sites on candidate probes and rank their specificity in regard to the underlying transcription factor. We developed a pipeline to analyze experimental data derived from compact universal protein binding microarrays and benchmarked it against two of the most accurate motif search methods. Our results indicate that MotifMark can be a viable alternative technique for prediction of motif from protein binding microarrays and possibly other related high-throughput techniques.
Fuzzy Constraints Linear Discriminant Analysis
Dec 30, 2016
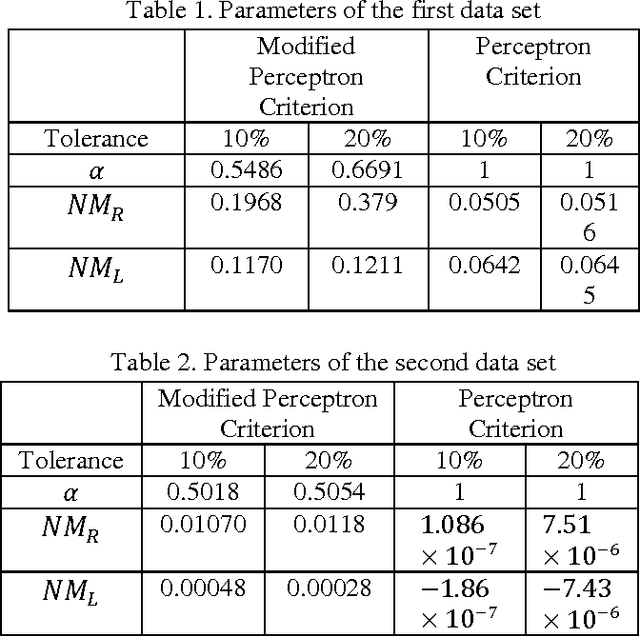
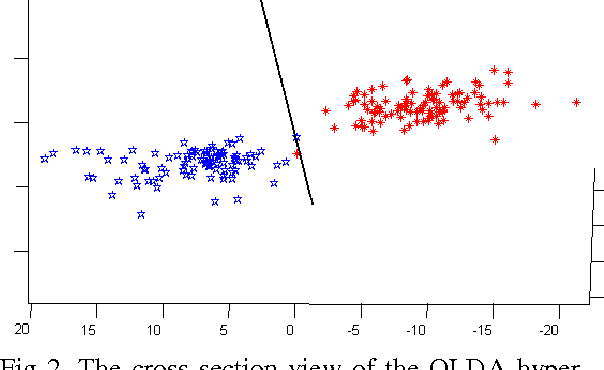
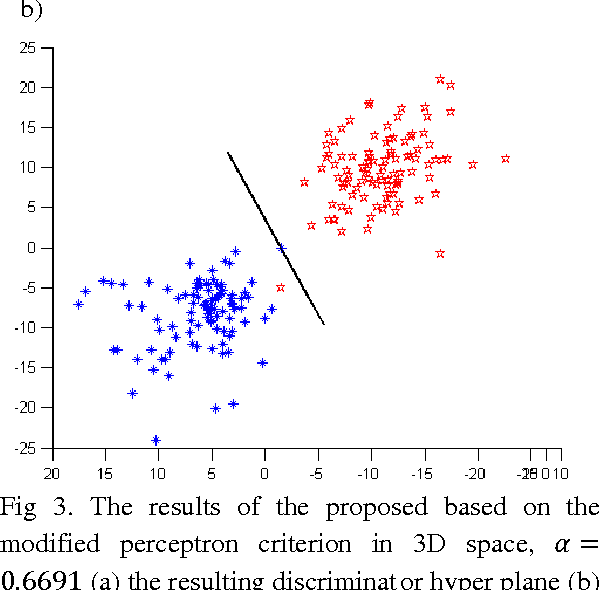
In this paper we introduce a fuzzy constraint linear discriminant analysis (FC-LDA). The FC-LDA tries to minimize misclassification error based on modified perceptron criterion that benefits handling the uncertainty near the decision boundary by means of a fuzzy linear programming approach with fuzzy resources. The method proposed has low computational complexity because of its linear characteristics and the ability to deal with noisy data with different degrees of tolerance. Obtained results verify the success of the algorithm when dealing with different problems. Comparing FC-LDA and LDA shows superiority in classification task.
A New Type-II Fuzzy Logic Based Controller for Non-linear Dynamical Systems with Application to a 3-PSP Parallel Robot
Dec 05, 2016
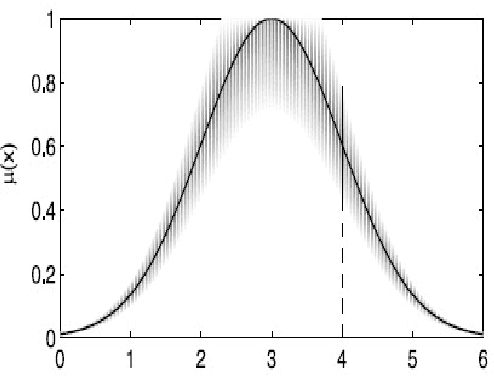

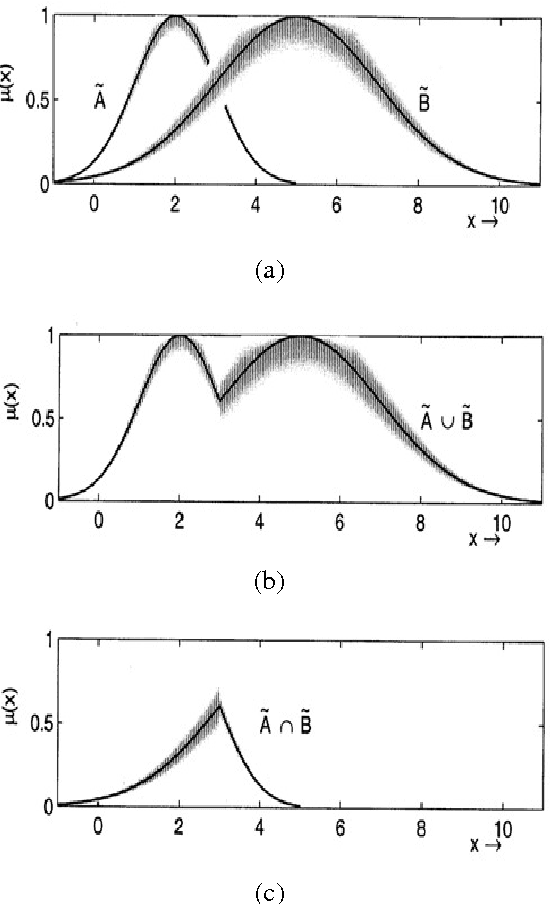
The concept of uncertainty is posed in almost any complex system including parallel robots as an outstanding instance of dynamical robotics systems. As suggested by the name, uncertainty, is some missing information that is beyond the knowledge of human thus we may tend to handle it properly to minimize the side-effects through the control process. Type-II fuzzy logic has shown its superiority over traditional fuzzy logic when dealing with uncertainty. Type-II fuzzy logic controllers are however newer and more promising approaches that have been recently applied to various fields due to their significant contribution especially when noise (as an important instance of uncertainty) emerges. During the design of Type-I fuzzy logic systems, we presume that we are almost certain about the fuzzy membership functions which is not true in many cases. Thus T2FLS as a more realistic approach dealing with practical applications might have a lot to offer. Type-II fuzzy logic takes into account a higher level of uncertainty, in other words, the membership grade for a type-II fuzzy variable is no longer a crisp number but rather is itself a type-I linguistic term. In this thesis the effects of uncertainty in dynamic control of a parallel robot is considered. More specifically, it is intended to incorporate the Type-II Fuzzy Logic paradigm into a model based controller, the so-called computed torque control method, and apply the result to a 3 degrees of freedom parallel manipulator. ...
DeeperBind: Enhancing Prediction of Sequence Specificities of DNA Binding Proteins
Nov 17, 2016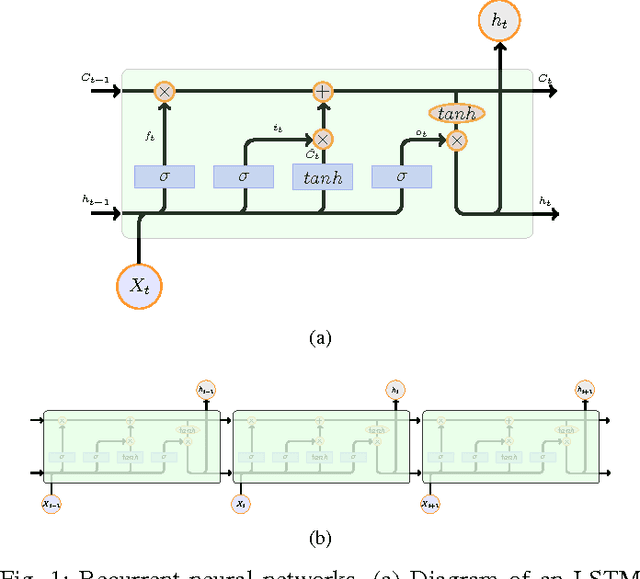


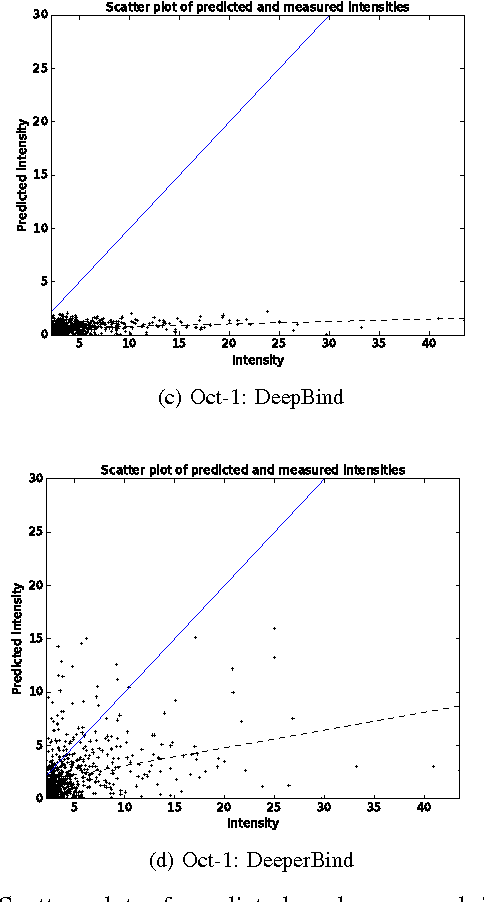
Transcription factors (TFs) are macromolecules that bind to \textit{cis}-regulatory specific sub-regions of DNA promoters and initiate transcription. Finding the exact location of these binding sites (aka motifs) is important in a variety of domains such as drug design and development. To address this need, several \textit{in vivo} and \textit{in vitro} techniques have been developed so far that try to characterize and predict the binding specificity of a protein to different DNA loci. The major problem with these techniques is that they are not accurate enough in prediction of the binding affinity and characterization of the corresponding motifs. As a result, downstream analysis is required to uncover the locations where proteins of interest bind. Here, we propose DeeperBind, a long short term recurrent convolutional network for prediction of protein binding specificities with respect to DNA probes. DeeperBind can model the positional dynamics of probe sequences and hence reckons with the contributions made by individual sub-regions in DNA sequences, in an effective way. Moreover, it can be trained and tested on datasets containing varying-length sequences. We apply our pipeline to the datasets derived from protein binding microarrays (PBMs), an in-vitro high-throughput technology for quantification of protein-DNA binding preferences, and present promising results. To the best of our knowledge, this is the most accurate pipeline that can predict binding specificities of DNA sequences from the data produced by high-throughput technologies through utilization of the power of deep learning for feature generation and positional dynamics modeling.
A Multi-Modal Graph-Based Semi-Supervised Pipeline for Predicting Cancer Survival
Nov 17, 2016



Cancer survival prediction is an active area of research that can help prevent unnecessary therapies and improve patient's quality of life. Gene expression profiling is being widely used in cancer studies to discover informative biomarkers that aid predict different clinical endpoint prediction. We use multiple modalities of data derived from RNA deep-sequencing (RNA-seq) to predict survival of cancer patients. Despite the wealth of information available in expression profiles of cancer tumors, fulfilling the aforementioned objective remains a big challenge, for the most part, due to the paucity of data samples compared to the high dimension of the expression profiles. As such, analysis of transcriptomic data modalities calls for state-of-the-art big-data analytics techniques that can maximally use all the available data to discover the relevant information hidden within a significant amount of noise. In this paper, we propose a pipeline that predicts cancer patients' survival by exploiting the structure of the input (manifold learning) and by leveraging the unlabeled samples using Laplacian support vector machines, a graph-based semi supervised learning (GSSL) paradigm. We show that under certain circumstances, no single modality per se will result in the best accuracy and by fusing different models together via a stacked generalization strategy, we may boost the accuracy synergistically. We apply our approach to two cancer datasets and present promising results. We maintain that a similar pipeline can be used for predictive tasks where labeled samples are expensive to acquire.