 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOne Set to Rule Them All: How to Obtain General Chemical Conditions via Bayesian Optimization over Curried Functions
Feb 26, 2025General parameters are highly desirable in the natural sciences - e.g., chemical reaction conditions that enable high yields across a range of related transformations. This has a significant practical impact since those general parameters can be transferred to related tasks without the need for laborious and time-intensive re-optimization. While Bayesian optimization (BO) is widely applied to find optimal parameter sets for specific tasks, it has remained underused in experiment planning towards such general optima. In this work, we consider the real-world problem of condition optimization for chemical reactions to study how performing generality-oriented BO can accelerate the identification of general optima, and whether these optima also translate to unseen examples. This is achieved through a careful formulation of the problem as an optimization over curried functions, as well as systematic evaluations of generality-oriented strategies for optimization tasks on real-world experimental data. We find that for generality-oriented optimization, simple myopic optimization strategies that decouple parameter and task selection perform comparably to more complex ones, and that effective optimization is merely determined by an effective exploration of both parameter and task space.
BoTier: Multi-Objective Bayesian Optimization with Tiered Composite Objectives
Jan 26, 2025
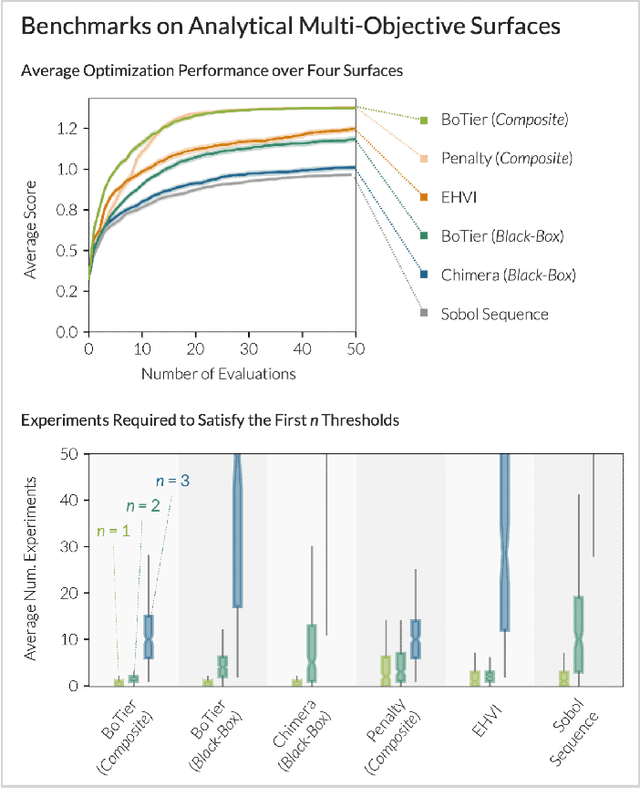
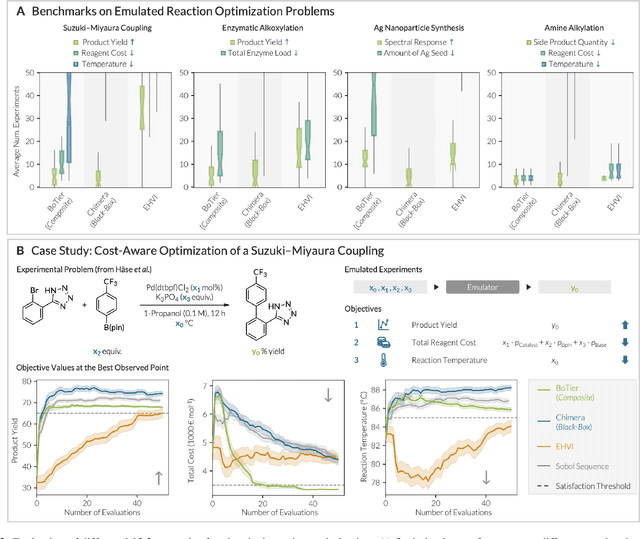
Scientific optimization problems are usually concerned with balancing multiple competing objectives, which come as preferences over both the outcomes of an experiment (e.g. maximize the reaction yield) and the corresponding input parameters (e.g. minimize the use of an expensive reagent). Typically, practical and economic considerations define a hierarchy over these objectives, which must be reflected in algorithms for sample-efficient experiment planning. Herein, we introduce BoTier, a composite objective that can flexibly represent a hierarchy of preferences over both experiment outcomes and input parameters. We provide systematic benchmarks on synthetic and real-life surfaces, demonstrating the robust applicability of BoTier across a number of use cases. Importantly, BoTier is implemented in an auto-differentiable fashion, enabling seamless integration with the BoTorch library, thereby facilitating adoption by the scientific community.
How to do impactful research in artificial intelligence for chemistry and materials science
Sep 16, 2024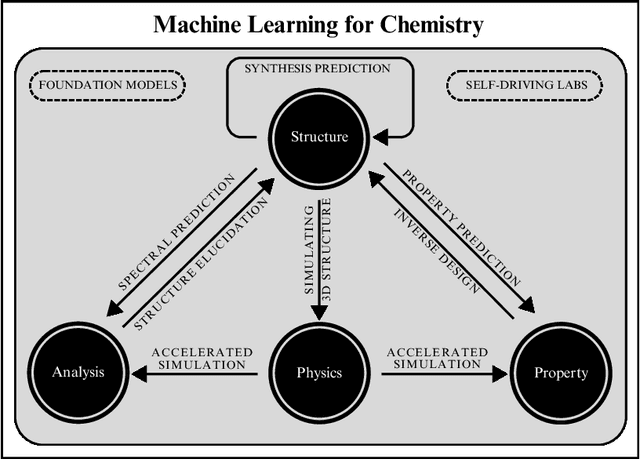
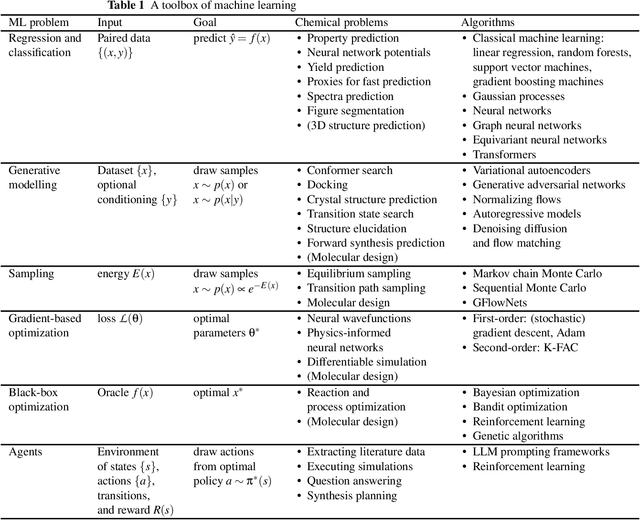
Machine learning has been pervasively touching many fields of science. Chemistry and materials science are no exception. While machine learning has been making a great impact, it is still not reaching its full potential or maturity. In this perspective, we first outline current applications across a diversity of problems in chemistry. Then, we discuss how machine learning researchers view and approach problems in the field. Finally, we provide our considerations for maximizing impact when researching machine learning for chemistry.
How Useful is Intermittent, Asynchronous Expert Feedback for Bayesian Optimization?
Jun 10, 2024



Bayesian optimization (BO) is an integral part of automated scientific discovery -- the so-called self-driving lab -- where human inputs are ideally minimal or at least non-blocking. However, scientists often have strong intuition, and thus human feedback is still useful. Nevertheless, prior works in enhancing BO with expert feedback, such as by incorporating it in an offline or online but blocking (arrives at each BO iteration) manner, are incompatible with the spirit of self-driving labs. In this work, we study whether a small amount of randomly arriving expert feedback that is being incorporated in a non-blocking manner can improve a BO campaign. To this end, we run an additional, independent computing thread on top of the BO loop to handle the feedback-gathering process. The gathered feedback is used to learn a Bayesian preference model that can readily be incorporated into the BO thread, to steer its exploration-exploitation process. Experiments on toy and chemistry datasets suggest that even just a few intermittent, asynchronous expert feedback can be useful for improving or constraining BO. This can especially be useful for its implication in improving self-driving labs, e.g. making them more data-efficient and less costly.
A Sober Look at LLMs for Material Discovery: Are They Actually Good for Bayesian Optimization Over Molecules?
Feb 07, 2024



Automation is one of the cornerstones of contemporary material discovery. Bayesian optimization (BO) is an essential part of such workflows, enabling scientists to leverage prior domain knowledge into efficient exploration of a large molecular space. While such prior knowledge can take many forms, there has been significant fanfare around the ancillary scientific knowledge encapsulated in large language models (LLMs). However, existing work thus far has only explored LLMs for heuristic materials searches. Indeed, recent work obtains the uncertainty estimate -- an integral part of BO -- from point-estimated, non-Bayesian LLMs. In this work, we study the question of whether LLMs are actually useful to accelerate principled Bayesian optimization in the molecular space. We take a sober, dispassionate stance in answering this question. This is done by carefully (i) viewing LLMs as fixed feature extractors for standard but principled BO surrogate models and by (ii) leveraging parameter-efficient finetuning methods and Bayesian neural networks to obtain the posterior of the LLM surrogate. Our extensive experiments with real-world chemistry problems show that LLMs can be useful for BO over molecules, but only if they have been pretrained or finetuned with domain-specific data.
SELFIES and the future of molecular string representations
Mar 31, 2022
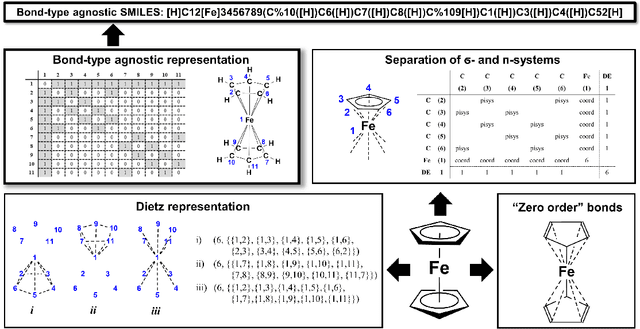
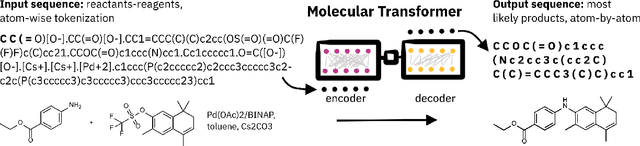
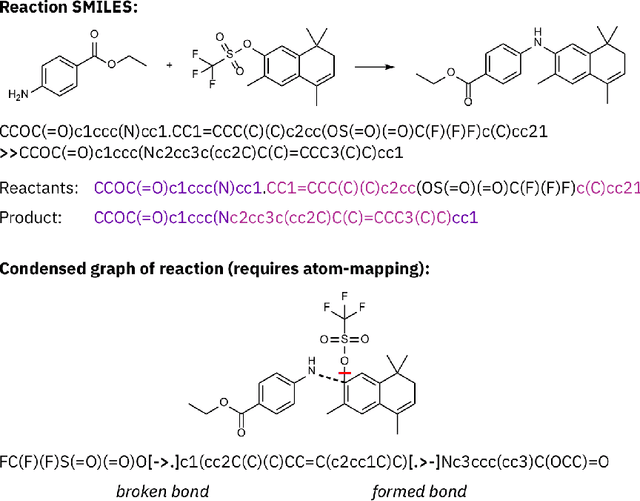
Artificial intelligence (AI) and machine learning (ML) are expanding in popularity for broad applications to challenging tasks in chemistry and materials science. Examples include the prediction of properties, the discovery of new reaction pathways, or the design of new molecules. The machine needs to read and write fluently in a chemical language for each of these tasks. Strings are a common tool to represent molecular graphs, and the most popular molecular string representation, SMILES, has powered cheminformatics since the late 1980s. However, in the context of AI and ML in chemistry, SMILES has several shortcomings -- most pertinently, most combinations of symbols lead to invalid results with no valid chemical interpretation. To overcome this issue, a new language for molecules was introduced in 2020 that guarantees 100\% robustness: SELFIES (SELF-referencIng Embedded Strings). SELFIES has since simplified and enabled numerous new applications in chemistry. In this manuscript, we look to the future and discuss molecular string representations, along with their respective opportunities and challenges. We propose 16 concrete Future Projects for robust molecular representations. These involve the extension toward new chemical domains, exciting questions at the interface of AI and robust languages and interpretability for both humans and machines. We hope that these proposals will inspire several follow-up works exploiting the full potential of molecular string representations for the future of AI in chemistry and materials science.