 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFew-step Cofolding with All-Atom Flow Maps
Jun 07, 2026All-atom generative modeling of 3D biomolecular complexes has emerged as the dominant paradigm for predicting the structure of proteins and protein-ligand systems. Generating structures at the atomic level of fidelity, however, typically requires expensive iterative diffusion rollouts, making both conventional deployment and inference-time search techniques computationally costly. In this paper, we introduce the Denoiser Cofolding All-Atom Flowmap (DeCAF) framework for distilling state-of-the-art all-atom cofolding models into all-atom flow maps that produce high-quality samples in only a few inference steps. We build DeCAF on a denoiser-based formulation of flow maps with endpoint losses that naturally support SE(3) rigid alignment, which we show is critical for training accurate models. We further derive a simple change of variables that lets DeCAF operate in the σ-space noise schedule of EDM-style architectures, enabling direct distillation from pretrained cofolding diffusion models. Equipped with DeCAF's flowmap lookahead, we introduce a purpose-built inference-time framework that improves sampling through reward-guided search. Empirically, DeCAF-Boltz statistically improves over Boltz-1x in both accuracy (RMSD) and physical validity scores of protein-ligand poses at strict NFE budgets on the challenging Runs N' Poses, while also showing a more optimal Pareto frontier across all inference compute budgets on PoseBusters. Distilling the state-of-the-art Pearl cofolding model, DeCAF-Pearl outperforms diffusion-based cofolding models and matches its teacher on success rate while using 5x fewer NFEs. We release our code at https://github.com/genesistherapeutics/decaf.
Learned Relay Representations for Forward-Thinking Discrete Diffusion Models
May 21, 2026When Masked Diffusion Models (MDMs) generate sequences through iterative refinement, the rich internal computation over masked positions is discarded, forcing every subsequent refinement step to recompute the valuable internal information stored as model representations. To avoid a hard reset between denoising rounds, we propose Learned Relay Representations (Relay), a method that allows MDMs to be forward-thinking when denoising by explicitly learning how to propagate latent information for the benefit of future denoising steps. Relay introduces a differentiable per-token channel that passes information between forward passes and is trained via truncated backpropagation through time (BPTT). We show that this framework can be scaled to state-of-the-art Diffusion Language Models (DLMs), and is seamlessly compatible with techniques like block diffusion and KV caching. We first provide a thorough justification of the design choices in Relay on a challenging Sudoku-based planning task. We then scale Relay to Fast-dLLM v2, a state-of-the-art DLM, outperforming standard supervised finetuning on coding tasks while reducing inference latency by up to 32%. Our empirical results demonstrate that state-of-the-art DLMs can be explicitly trained to relay latent information forward across decoding steps, advancing the performance-latency Pareto frontier. We provide code for all our experiments.
Aligning Flow Map Policies with Optimal Q-Guidance
May 12, 2026Generative policies based on expressive model classes, such as diffusion and flow matching, are well-suited to complex control problems with highly multimodal action distributions. Their expressivity, however, comes at a significant inference cost: generating each action typically requires simulating many steps of the generative process, compounding latency across sequential decision-making rollouts. We introduce flow map policies, a novel class of generative policies designed for fast action generation by learning to take arbitrary-size jumps including one-step jumps-across the generative dynamics of existing flow-based policies. We instantiate flow map policies for offline-to-online reinforcement learning (RL) and formulate online adaptation as a trust-region optimization problem that improves the critic's Q-value while remaining close to the offline policy. We theoretically derive FLOW MAP Q-GUIDANCE (FMQ), a principled closed-form learning target that is optimal for adapting offline flow map policies under a critic-guided trust-region constraint. We further introduce Q-GUIDED BEAM SEARCH (QGBS), a stochastic flow-map sampler that combines renoising with beam search to enable iterative inference-time refinement. Across 12 challenging robotic manipulation and locomotion tasks from OGBench and RoboMimic, FMQ achieves state-of-the-art performance in offline-to-online RL, outperforming the previous one-step policy MVP by a relative improvement of 21.3% on the average success rate.
General Multimodal Protein Design Enables DNA-Encoding of Chemistry
Apr 06, 2026Evolution is an extraordinary engine for enzymatic diversity, yet the chemistry it has explored remains a narrow slice of what DNA can encode. Deep generative models can design new proteins that bind ligands, but none have created enzymes without pre-specifying catalytic residues. We introduce DISCO (DIffusion for Sequence-structure CO-design), a multimodal model that co-designs protein sequence and 3D structure around arbitrary biomolecules, as well as inference-time scaling methods that optimize objectives across both modalities. Conditioned solely on reactive intermediates, DISCO designs diverse heme enzymes with novel active-site geometries. These enzymes catalyze new-to-nature carbene-transfer reactions, including alkene cyclopropanation, spirocyclopropanation, B-H, and C(sp$^3$)-H insertions, with high activities exceeding those of engineered enzymes. Random mutagenesis of a selected design further confirmed that enzyme activity can be improved through directed evolution. By providing a scalable route to evolvable enzymes, DISCO broadens the potential scope of genetically encodable transformations. Code is available at https://github.com/DISCO-design/DISCO.
Curly Flow Matching for Learning Non-gradient Field Dynamics
Oct 30, 2025Modeling the transport dynamics of natural processes from population-level observations is a ubiquitous problem in the natural sciences. Such models rely on key assumptions about the underlying process in order to enable faithful learning of governing dynamics that mimic the actual system behavior. The de facto assumption in current approaches relies on the principle of least action that results in gradient field dynamics and leads to trajectories minimizing an energy functional between two probability measures. However, many real-world systems, such as cell cycles in single-cell RNA, are known to exhibit non-gradient, periodic behavior, which fundamentally cannot be captured by current state-of-the-art methods such as flow and bridge matching. In this paper, we introduce Curly Flow Matching (Curly-FM), a novel approach that is capable of learning non-gradient field dynamics by designing and solving a Schr\"odinger bridge problem with a non-zero drift reference process -- in stark contrast to typical zero-drift reference processes -- which is constructed using inferred velocities in addition to population snapshot data. We showcase Curly-FM by solving the trajectory inference problems for single cells, computational fluid dynamics, and ocean currents with approximate velocities. We demonstrate that Curly-FM can learn trajectories that better match both the reference process and population marginals. Curly-FM expands flow matching models beyond the modeling of populations and towards the modeling of known periodic behavior in physical systems. Our code repository is accessible at: https://github.com/kpetrovicc/curly-flow-matching.git
Generalised Flow Maps for Few-Step Generative Modelling on Riemannian Manifolds
Oct 24, 2025Geometric data and purpose-built generative models on them have become ubiquitous in high-impact deep learning application domains, ranging from protein backbone generation and computational chemistry to geospatial data. Current geometric generative models remain computationally expensive at inference -- requiring many steps of complex numerical simulation -- as they are derived from dynamical measure transport frameworks such as diffusion and flow-matching on Riemannian manifolds. In this paper, we propose Generalised Flow Maps (GFM), a new class of few-step generative models that generalises the Flow Map framework in Euclidean spaces to arbitrary Riemannian manifolds. We instantiate GFMs with three self-distillation-based training methods: Generalised Lagrangian Flow Maps, Generalised Eulerian Flow Maps, and Generalised Progressive Flow Maps. We theoretically show that GFMs, under specific design decisions, unify and elevate existing Euclidean few-step generative models, such as consistency models, shortcut models, and meanflows, to the Riemannian setting. We benchmark GFMs against other geometric generative models on a suite of geometric datasets, including geospatial data, RNA torsion angles, and hyperbolic manifolds, and achieve state-of-the-art sample quality for single- and few-step evaluations, and superior or competitive log-likelihoods using the implicit probability flow.
RETRO SYNFLOW: Discrete Flow Matching for Accurate and Diverse Single-Step Retrosynthesis
Jun 04, 2025A fundamental problem in organic chemistry is identifying and predicting the series of reactions that synthesize a desired target product molecule. Due to the combinatorial nature of the chemical search space, single-step reactant prediction -- i.e. single-step retrosynthesis -- remains challenging even for existing state-of-the-art template-free generative approaches to produce an accurate yet diverse set of feasible reactions. In this paper, we model single-step retrosynthesis planning and introduce RETRO SYNFLOW (RSF) a discrete flow-matching framework that builds a Markov bridge between the prescribed target product molecule and the reactant molecule. In contrast to past approaches, RSF employs a reaction center identification step to produce intermediate structures known as synthons as a more informative source distribution for the discrete flow. To further enhance diversity and feasibility of generated samples, we employ Feynman-Kac steering with Sequential Monte Carlo based resampling to steer promising generations at inference using a new reward oracle that relies on a forward-synthesis model. Empirically, we demonstrate \nameshort achieves $60.0 \%$ top-1 accuracy, which outperforms the previous SOTA by $20 \%$. We also substantiate the benefits of steering at inference and demonstrate that FK-steering improves top-$5$ round-trip accuracy by $19 \%$ over prior template-free SOTA methods, all while preserving competitive top-$k$ accuracy results.
Scalable Equilibrium Sampling with Sequential Boltzmann Generators
Feb 25, 2025

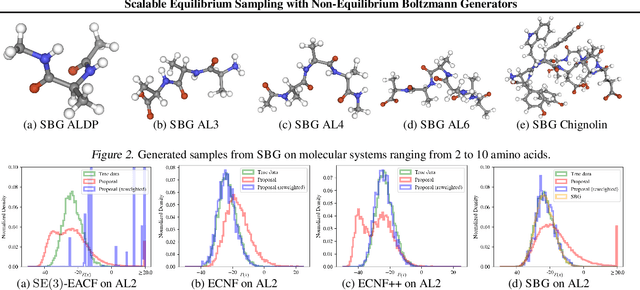

Scalable sampling of molecular states in thermodynamic equilibrium is a long-standing challenge in statistical physics. Boltzmann generators tackle this problem by pairing powerful normalizing flows with importance sampling to obtain statistically independent samples under the target distribution. In this paper, we extend the Boltzmann generator framework and introduce Sequential Boltzmann generators (SBG) with two key improvements. The first is a highly efficient non-equivariant Transformer-based normalizing flow operating directly on all-atom Cartesian coordinates. In contrast to equivariant continuous flows of prior methods, we leverage exactly invertible non-equivariant architectures which are highly efficient both during sample generation and likelihood computation. As a result, this unlocks more sophisticated inference strategies beyond standard importance sampling. More precisely, as a second key improvement we perform inference-time scaling of flow samples using annealed Langevin dynamics which transports samples toward the target distribution leading to lower variance (annealed) importance weights which enable higher fidelity resampling with sequential Monte Carlo. SBG achieves state-of-the-art performance w.r.t. all metrics on molecular systems, demonstrating the first equilibrium sampling in Cartesian coordinates of tri, tetra, and hexapeptides that were so far intractable for prior Boltzmann generators.
The Superposition of Diffusion Models Using the Itô Density Estimator
Dec 23, 2024The Cambrian explosion of easily accessible pre-trained diffusion models suggests a demand for methods that combine multiple different pre-trained diffusion models without incurring the significant computational burden of re-training a larger combined model. In this paper, we cast the problem of combining multiple pre-trained diffusion models at the generation stage under a novel proposed framework termed superposition. Theoretically, we derive superposition from rigorous first principles stemming from the celebrated continuity equation and design two novel algorithms tailor-made for combining diffusion models in SuperDiff. SuperDiff leverages a new scalable It\^o density estimator for the log likelihood of the diffusion SDE which incurs no additional overhead compared to the well-known Hutchinson's estimator needed for divergence calculations. We demonstrate that SuperDiff is scalable to large pre-trained diffusion models as superposition is performed solely through composition during inference, and also enjoys painless implementation as it combines different pre-trained vector fields through an automated re-weighting scheme. Notably, we show that SuperDiff is efficient during inference time, and mimics traditional composition operators such as the logical OR and the logical AND. We empirically demonstrate the utility of using SuperDiff for generating more diverse images on CIFAR-10, more faithful prompt conditioned image editing using Stable Diffusion, and improved unconditional de novo structure design of proteins. https://github.com/necludov/super-diffusion
Steering Masked Discrete Diffusion Models via Discrete Denoising Posterior Prediction
Oct 10, 2024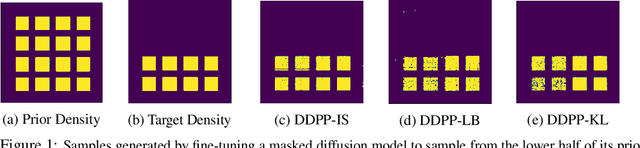

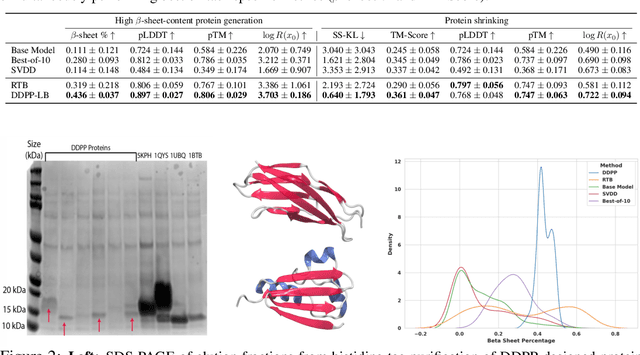

Generative modeling of discrete data underlies important applications spanning text-based agents like ChatGPT to the design of the very building blocks of life in protein sequences. However, application domains need to exert control over the generated data by steering the generative process - typically via RLHF - to satisfy a specified property, reward, or affinity metric. In this paper, we study the problem of steering Masked Diffusion Models (MDMs), a recent class of discrete diffusion models that offer a compelling alternative to traditional autoregressive models. We introduce Discrete Denoising Posterior Prediction (DDPP), a novel framework that casts the task of steering pre-trained MDMs as a problem of probabilistic inference by learning to sample from a target Bayesian posterior. Our DDPP framework leads to a family of three novel objectives that are all simulation-free, and thus scalable while applying to general non-differentiable reward functions. Empirically, we instantiate DDPP by steering MDMs to perform class-conditional pixel-level image modeling, RLHF-based alignment of MDMs using text-based rewards, and finetuning protein language models to generate more diverse secondary structures and shorter proteins. We substantiate our designs via wet-lab validation, where we observe transient expression of reward-optimized protein sequences.