 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssessing the impacts of tradable credit schemes through agent-based simulation
Feb 17, 2025Tradable credit schemes (TCS) have been attracting interest from the transportation research community as an appealing alternative to congestion pricing, due to the advantages of revenue neutrality and equity. Nonetheless, existing research has largely employed network and market equilibrium approaches with simplistic characterizations of transportation demand, supply, credit market operations, and market behavior. Agent- and activity-based simulation affords a natural means to comprehensively assess TCS by more realistically modeling demand, supply, and individual market interactions. We propose an integrated simulation framework for modeling a TCS, and implements it within the state-of-the-art open-source urban simulation platform SimMobility, including: (a) a flexible TCS design that considers multiple trips and explicitly accounts for individual trading behaviors; (b) a simulation framework that captures the complex interactions between a TCS regulator, the traveler, and the TCS market itself, with the flexibility to test future TCS designs and relevant mobility models; and (c) a set of simulation experiments on a large mesoscopic multimodal network combined with a Bayesian Optimization approach for TCS optimal design. The experiment results indicate network and market performance to stabilize over the day-to-day process, showing the alignment of our agent-based simulation with the known theoretical properties of TCS. We confirm the efficiency of TCS in reducing congestion under the adopted market behavioral assumptions and open the door for simulating different individual behaviors. We measure how TCS impacts differently the local network, heterogeneous users, the different travel behaviors, and how testing different TCS designs can avoid negative market trading behaviors.
Graph Positional and Structural Encoder
Jul 14, 2023



Positional and structural encodings (PSE) enable better identifiability of nodes within a graph, as in general graphs lack a canonical node ordering. This renders PSEs essential tools for empowering modern GNNs, and in particular graph Transformers. However, designing PSEs that work optimally for a variety of graph prediction tasks is a challenging and unsolved problem. Here, we present the graph positional and structural encoder (GPSE), a first-ever attempt to train a graph encoder that captures rich PSE representations for augmenting any GNN. GPSE can effectively learn a common latent representation for multiple PSEs, and is highly transferable. The encoder trained on a particular graph dataset can be used effectively on datasets drawn from significantly different distributions and even modalities. We show that across a wide range of benchmarks, GPSE-enhanced models can significantly improve the performance in certain tasks, while performing on par with those that employ explicitly computed PSEs in other cases. Our results pave the way for the development of large pre-trained models for extracting graph positional and structural information and highlight their potential as a viable alternative to explicitly computed PSEs as well as to existing self-supervised pre-training approaches.
Single-Cell Multimodal Prediction via Transformers
Mar 01, 2023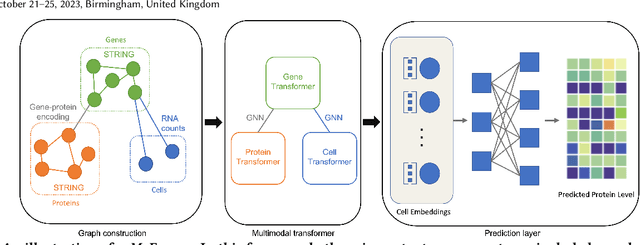
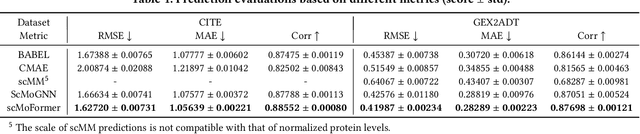
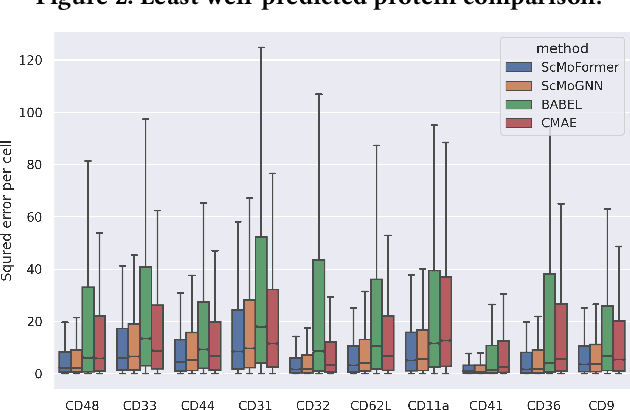
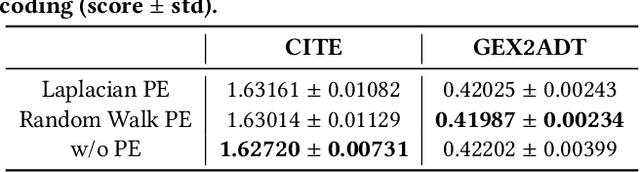
The recent development of multimodal single-cell technology has made the possibility of acquiring multiple omics data from individual cells, thereby enabling a deeper understanding of cellular states and dynamics. Nevertheless, the proliferation of multimodal single-cell data also introduces tremendous challenges in modeling the complex interactions among different modalities. The recently advanced methods focus on constructing static interaction graphs and applying graph neural networks (GNNs) to learn from multimodal data. However, such static graphs can be suboptimal as they do not take advantage of the downstream task information; meanwhile GNNs also have some inherent limitations when deeply stacking GNN layers. To tackle these issues, in this work, we investigate how to leverage transformers for multimodal single-cell data in an end-to-end manner while exploiting downstream task information. In particular, we propose a scMoFormer framework which can readily incorporate external domain knowledge and model the interactions within each modality and cross modalities. Extensive experiments demonstrate that scMoFormer achieves superior performance on various benchmark datasets. Note that scMoFormer won a Kaggle silver medal with the rank of $24\ /\ 1221$ (Top 2%) without ensemble in a NeurIPS 2022 competition. Our implementation is publicly available at Github.
Single Cells Are Spatial Tokens: Transformers for Spatial Transcriptomic Data Imputation
Feb 06, 2023
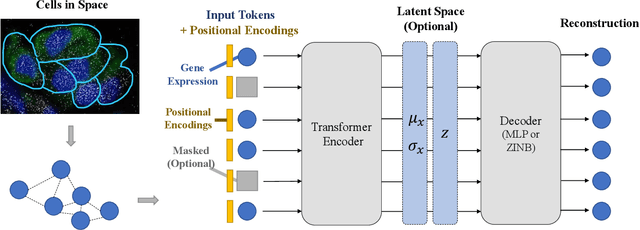
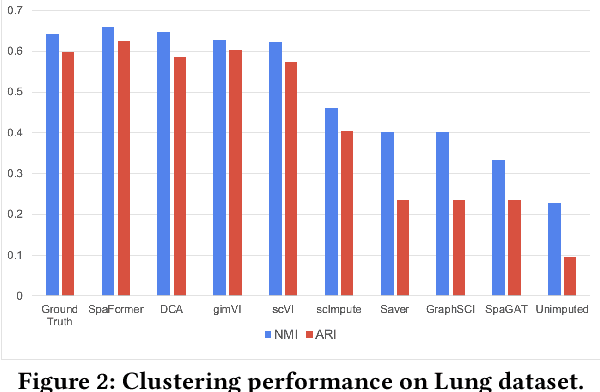
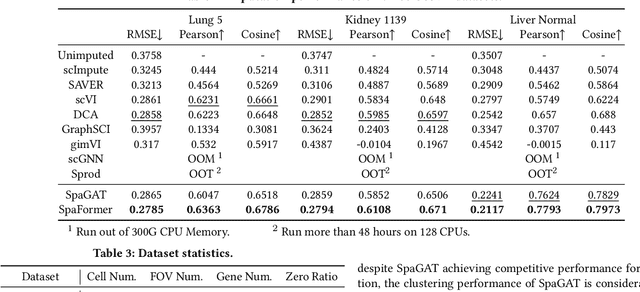
Spatially resolved transcriptomics brings exciting breakthroughs to single-cell analysis by providing physical locations along with gene expression. However, as a cost of the extremely high spatial resolution, the cellular level spatial transcriptomic data suffer significantly from missing values. While a standard solution is to perform imputation on the missing values, most existing methods either overlook spatial information or only incorporate localized spatial context without the ability to capture long-range spatial information. Using multi-head self-attention mechanisms and positional encoding, transformer models can readily grasp the relationship between tokens and encode location information. In this paper, by treating single cells as spatial tokens, we study how to leverage transformers to facilitate spatial tanscriptomics imputation. In particular, investigate the following two key questions: (1) $\textit{how to encode spatial information of cells in transformers}$, and (2) $\textit{ how to train a transformer for transcriptomic imputation}$. By answering these two questions, we present a transformer-based imputation framework, SpaFormer, for cellular-level spatial transcriptomic data. Extensive experiments demonstrate that SpaFormer outperforms existing state-of-the-art imputation algorithms on three large-scale datasets.
Taxonomy of Benchmarks in Graph Representation Learning
Jun 15, 2022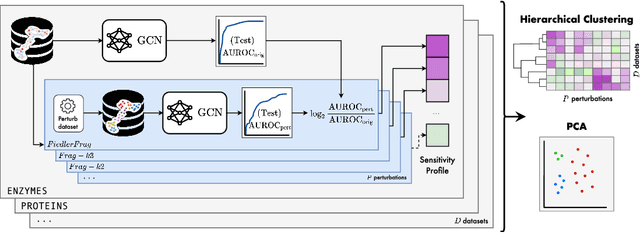
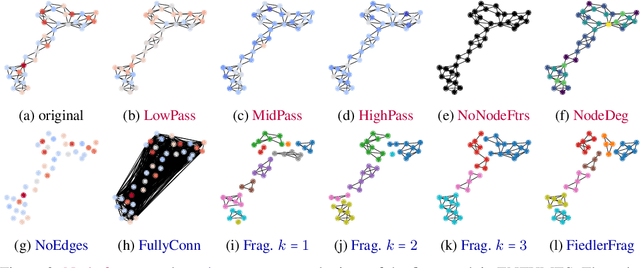
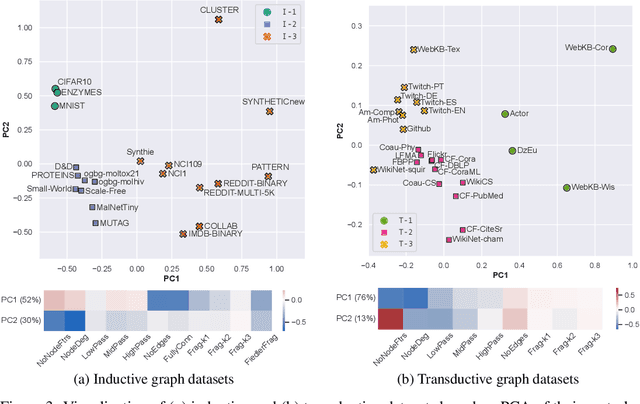
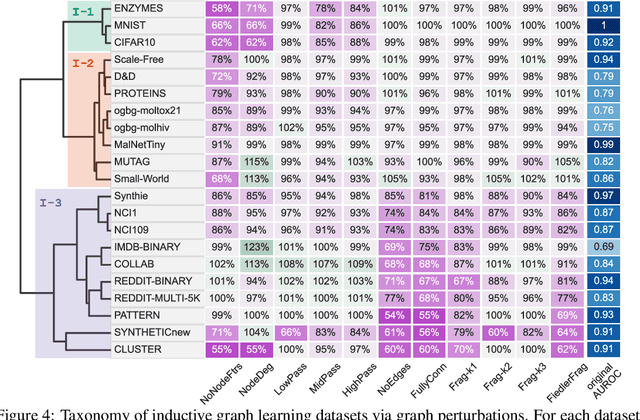
Graph Neural Networks (GNNs) extend the success of neural networks to graph-structured data by accounting for their intrinsic geometry. While extensive research has been done on developing GNN models with superior performance according to a collection of graph representation learning benchmarks, it is currently not well understood what aspects of a given model are probed by them. For example, to what extent do they test the ability of a model to leverage graph structure vs. node features? Here, we develop a principled approach to taxonomize benchmarking datasets according to a $\textit{sensitivity profile}$ that is based on how much GNN performance changes due to a collection of graph perturbations. Our data-driven analysis provides a deeper understanding of which benchmarking data characteristics are leveraged by GNNs. Consequently, our taxonomy can aid in selection and development of adequate graph benchmarks, and better informed evaluation of future GNN methods. Finally, our approach and implementation in $\texttt{GTaxoGym}$ package are extendable to multiple graph prediction task types and future datasets.
Towards a Taxonomy of Graph Learning Datasets
Oct 27, 2021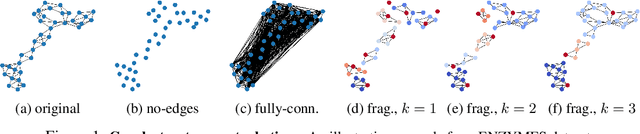
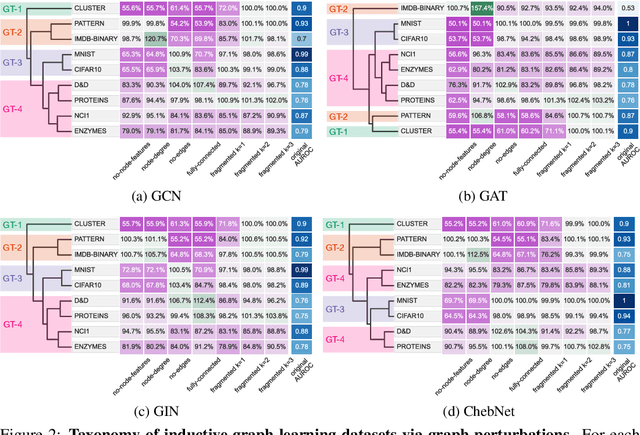
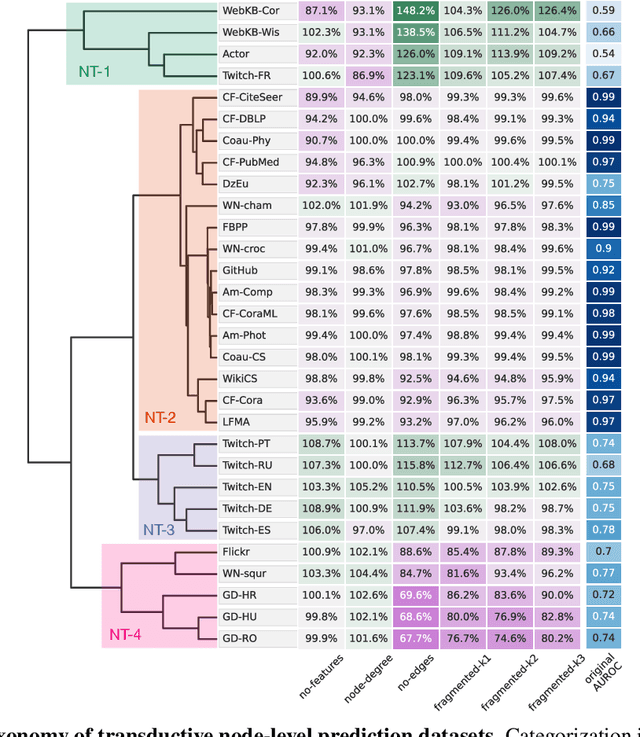
Graph neural networks (GNNs) have attracted much attention due to their ability to leverage the intrinsic geometries of the underlying data. Although many different types of GNN models have been developed, with many benchmarking procedures to demonstrate the superiority of one GNN model over the others, there is a lack of systematic understanding of the underlying benchmarking datasets, and what aspects of the model are being tested. Here, we provide a principled approach to taxonomize graph benchmarking datasets by carefully designing a collection of graph perturbations to probe the essential data characteristics that GNN models leverage to perform predictions. Our data-driven taxonomization of graph datasets provides a new understanding of critical dataset characteristics that will enable better model evaluation and the development of more specialized GNN models.
Accurately Modeling Biased Random Walks on Weighted Graphs Using $\textit{Node2vec+}$
Sep 15, 2021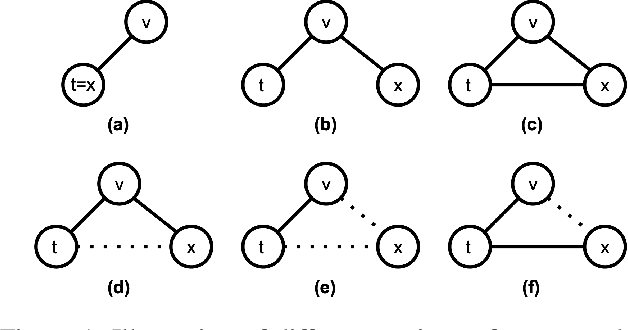
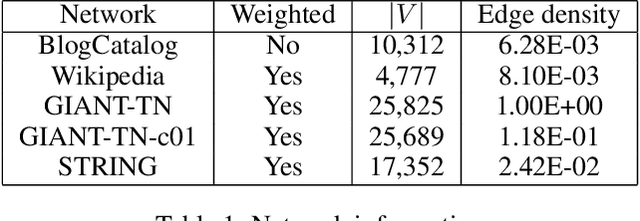
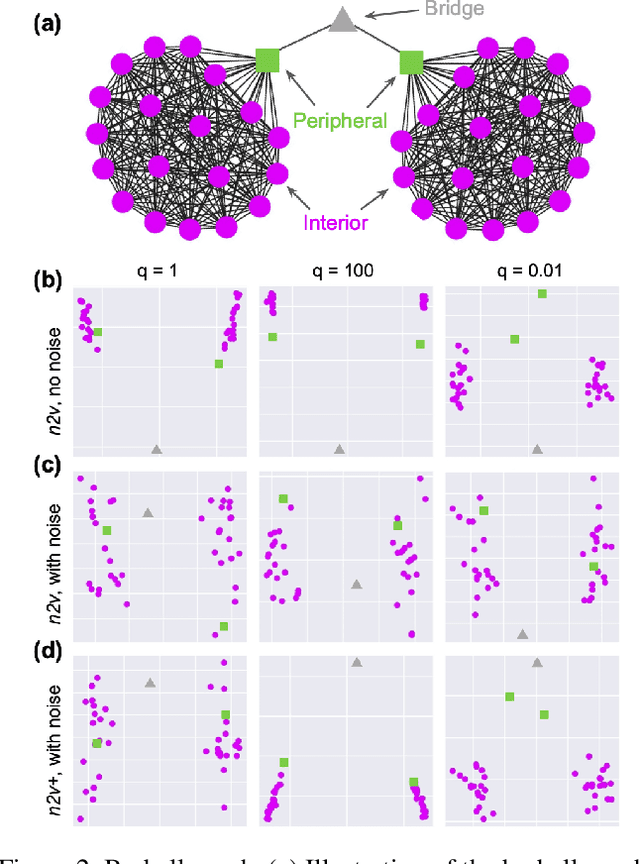
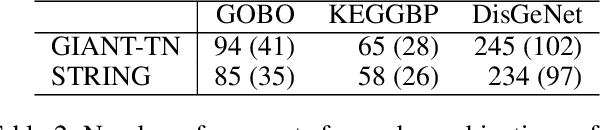
Node embedding is a powerful approach for representing the structural role of each node in a graph. $\textit{Node2vec}$ is a widely used method for node embedding that works by exploring the local neighborhoods via biased random walks on the graph. However, $\textit{node2vec}$ does not consider edge weights when computing walk biases. This intrinsic limitation prevents $\textit{node2vec}$ from leveraging all the information in weighted graphs and, in turn, limits its application to many real-world networks that are weighted and dense. Here, we naturally extend $\textit{node2vec}$ to $\textit{node2vec+}$ in a way that accounts for edge weights when calculating walk biases, but which reduces to $\textit{node2vec}$ in the cases of unweighted graphs or unbiased walks. We empirically show that $\textit{node2vec+}$ is more robust to additive noise than $\textit{node2vec}$ in weighted graphs using two synthetic datasets. We also demonstrate that $\textit{node2vec+}$ significantly outperforms $\textit{node2vec}$ on a commonly benchmarked multi-label dataset (Wikipedia). Furthermore, we test $\textit{node2vec+}$ against GCN and GraphSAGE using various challenging gene classification tasks on two protein-protein interaction networks. Despite some clear advantages of GCN and GraphSAGE, they show comparable performance with $\textit{node2vec+}$. Finally, $\textit{node2vec+}$ can be used as a general approach for generating biased random walks, benefiting all existing methods built on top of $\textit{node2vec}$. $\textit{Node2vec+}$ is implemented as part of $\texttt{PecanPy}$, which is available at https://github.com/krishnanlab/PecanPy .