 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMSP-LLM: A Unified Large Language Model Framework for Complete Material Synthesis Planning
Feb 07, 2026Material synthesis planning (MSP) remains a fundamental and underexplored bottleneck in AI-driven materials discovery, as it requires not only identifying suitable precursor materials but also designing coherent sequences of synthesis operations to realize a target material. Although several AI-based approaches have been proposed to address isolated subtasks of MSP, a unified methodology for solving the entire MSP task has yet to be established. We propose MSP-LLM, a unified LLM-based framework that formulates MSP as a structured process composed of two constituent subproblems: precursor prediction (PP) and synthesis operation prediction (SOP). Our approach introduces a discrete material class as an intermediate decision variable that organizes both tasks into a chemically consistent decision chain. For OP, we further incorporate hierarchical precursor types as synthesis-relevant inductive biases and employ an explicit conditioning strategy that preserves precursor-related information in the autoregressive decoding state. Extensive experiments show that MSP-LLM consistently outperforms existing methods on both PP and SOP, as well as on the complete MSP task, demonstrating an effective and scalable framework for MSP that can accelerate real-world materials discovery.
Thickness-aware E(3)-Equivariant 3D Mesh Neural Networks
May 27, 2025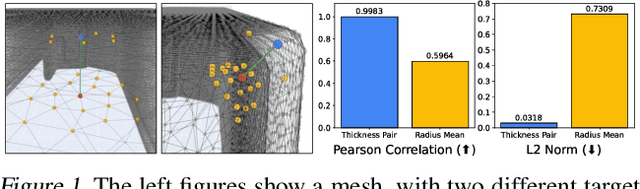
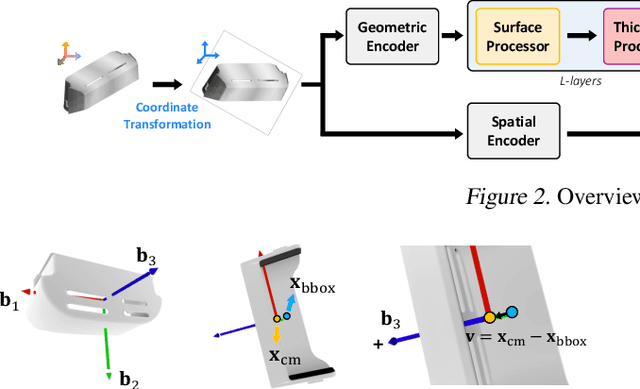
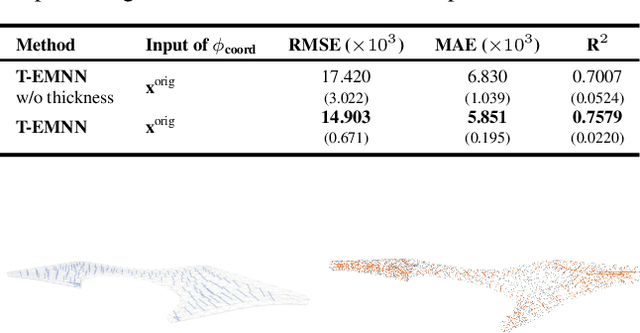

Mesh-based 3D static analysis methods have recently emerged as efficient alternatives to traditional computational numerical solvers, significantly reducing computational costs and runtime for various physics-based analyses. However, these methods primarily focus on surface topology and geometry, often overlooking the inherent thickness of real-world 3D objects, which exhibits high correlations and similar behavior between opposing surfaces. This limitation arises from the disconnected nature of these surfaces and the absence of internal edge connections within the mesh. In this work, we propose a novel framework, the Thickness-aware E(3)-Equivariant 3D Mesh Neural Network (T-EMNN), that effectively integrates the thickness of 3D objects while maintaining the computational efficiency of surface meshes. Additionally, we introduce data-driven coordinates that encode spatial information while preserving E(3)-equivariance or invariance properties, ensuring consistent and robust analysis. Evaluations on a real-world industrial dataset demonstrate the superior performance of T-EMNN in accurately predicting node-level 3D deformations, effectively capturing thickness effects while maintaining computational efficiency.
MedRep: Medical Concept Representation for General Electronic Health Record Foundation Models
Apr 11, 2025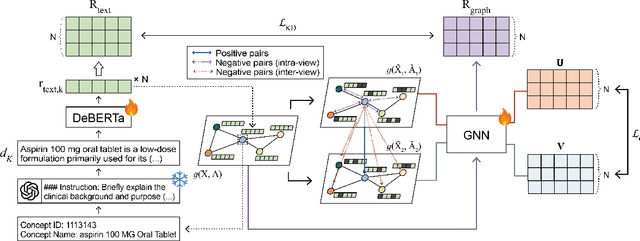
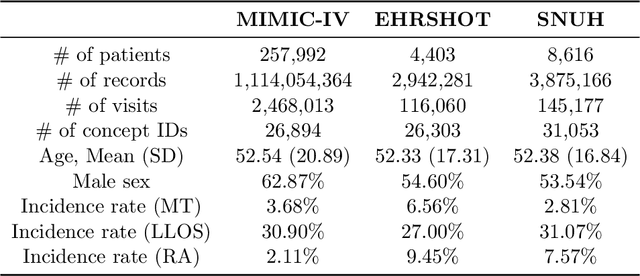
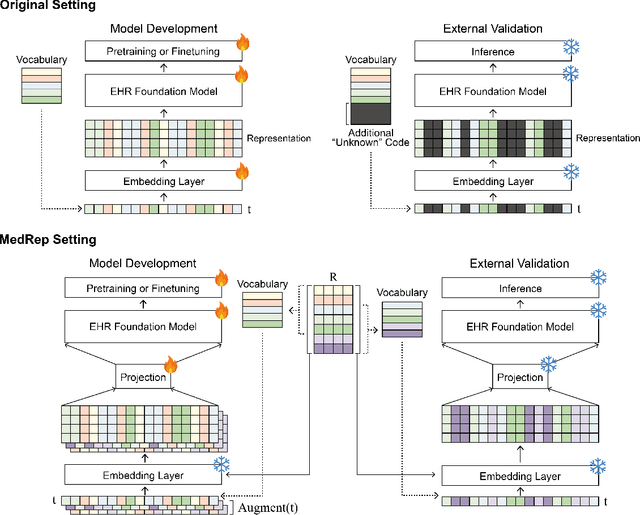

Electronic health record (EHR) foundation models have been an area ripe for exploration with their improved performance in various medical tasks. Despite the rapid advances, there exists a fundamental limitation: Processing unseen medical codes out of the vocabulary. This problem limits the generality of EHR foundation models and the integration of models trained with different vocabularies. To deal with this problem, we propose MedRep for EHR foundation models based on the observational medical outcome partnership (OMOP) common data model (CDM), providing the integrated medical concept representations and the basic data augmentation strategy for patient trajectories. For concept representation learning, we enrich the information of each concept with a minimal definition through large language model (LLM) prompts and enhance the text-based representations through graph ontology of OMOP vocabulary. Trajectory augmentation randomly replaces selected concepts with other similar concepts that have closely related representations to let the model practice with the concepts out-of-vocabulary. Finally, we demonstrate that EHR foundation models trained with MedRep better maintain the prediction performance in external datasets. Our code implementation is publicly available at https://github.com/kicarussays/MedRep.
Subgraph Federated Learning for Local Generalization
Mar 06, 2025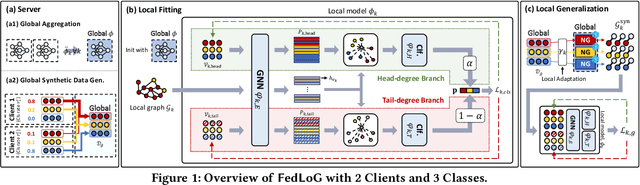
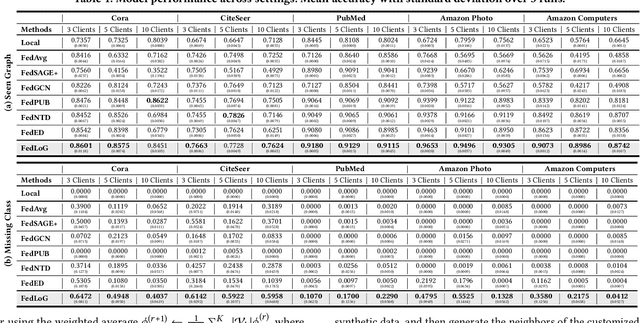
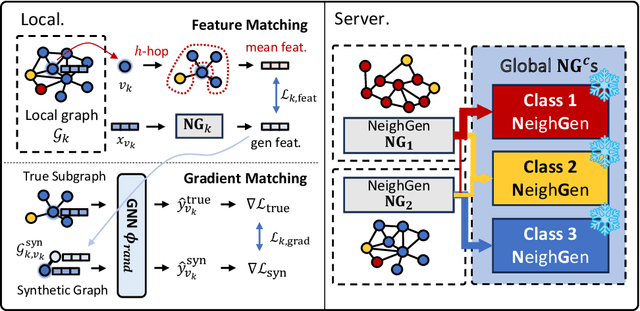
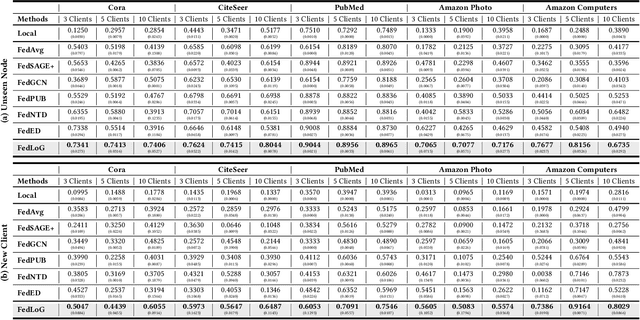
Federated Learning (FL) on graphs enables collaborative model training to enhance performance without compromising the privacy of each client. However, existing methods often overlook the mutable nature of graph data, which frequently introduces new nodes and leads to shifts in label distribution. Since they focus solely on performing well on each client's local data, they are prone to overfitting to their local distributions (i.e., local overfitting), which hinders their ability to generalize to unseen data with diverse label distributions. In contrast, our proposed method, FedLoG, effectively tackles this issue by mitigating local overfitting. Our model generates global synthetic data by condensing the reliable information from each class representation and its structural information across clients. Using these synthetic data as a training set, we alleviate the local overfitting problem by adaptively generalizing the absent knowledge within each local dataset. This enhances the generalization capabilities of local models, enabling them to handle unseen data effectively. Our model outperforms baselines in our proposed experimental settings, which are designed to measure generalization power to unseen data in practical scenarios. Our code is available at https://github.com/sung-won-kim/FedLoG
RAG-Enhanced Collaborative LLM Agents for Drug Discovery
Feb 22, 2025Recent advances in large language models (LLMs) have shown great potential to accelerate drug discovery. However, the specialized nature of biochemical data often necessitates costly domain-specific fine-tuning, posing critical challenges. First, it hinders the application of more flexible general-purpose LLMs in cutting-edge drug discovery tasks. More importantly, it impedes the rapid integration of the vast amounts of scientific data continuously generated through experiments and research. To investigate these challenges, we propose CLADD, a retrieval-augmented generation (RAG)-empowered agentic system tailored to drug discovery tasks. Through the collaboration of multiple LLM agents, CLADD dynamically retrieves information from biomedical knowledge bases, contextualizes query molecules, and integrates relevant evidence to generate responses -- all without the need for domain-specific fine-tuning. Crucially, we tackle key obstacles in applying RAG workflows to biochemical data, including data heterogeneity, ambiguity, and multi-source integration. We demonstrate the flexibility and effectiveness of this framework across a variety of drug discovery tasks, showing that it outperforms general-purpose and domain-specific LLMs as well as traditional deep learning approaches.
3D Interaction Geometric Pre-training for Molecular Relational Learning
Dec 04, 2024Molecular Relational Learning (MRL) is a rapidly growing field that focuses on understanding the interaction dynamics between molecules, which is crucial for applications ranging from catalyst engineering to drug discovery. Despite recent progress, earlier MRL approaches are limited to using only the 2D topological structure of molecules, as obtaining the 3D interaction geometry remains prohibitively expensive. This paper introduces a novel 3D geometric pre-training strategy for MRL (3DMRL) that incorporates a 3D virtual interaction environment, overcoming the limitations of costly traditional quantum mechanical calculation methods. With the constructed 3D virtual interaction environment, 3DMRL trains 2D MRL model to learn the overall 3D geometric information of molecular interaction through contrastive learning. Moreover, fine-grained interaction between molecules is learned through force prediction loss, which is crucial in understanding the wide range of molecular interaction processes. Extensive experiments on various tasks using real-world datasets, including out-of-distribution and extrapolation scenarios, demonstrate the effectiveness of 3DMRL, showing up to a 24.93\% improvement in performance across 40 tasks.
Retrieval-Retro: Retrieval-based Inorganic Retrosynthesis with Expert Knowledge
Oct 28, 2024
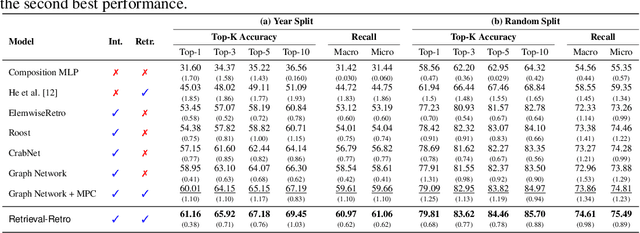
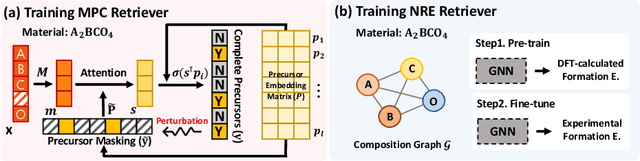
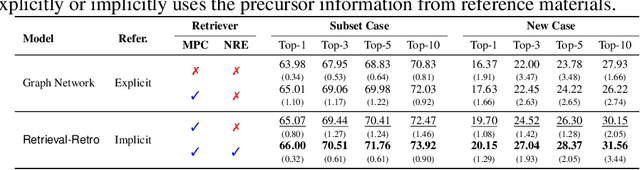
While inorganic retrosynthesis planning is essential in the field of chemical science, the application of machine learning in this area has been notably less explored compared to organic retrosynthesis planning. In this paper, we propose Retrieval-Retro for inorganic retrosynthesis planning, which implicitly extracts the precursor information of reference materials that are retrieved from the knowledge base regarding domain expertise in the field. Specifically, instead of directly employing the precursor information of reference materials, we propose implicitly extracting it with various attention layers, which enables the model to learn novel synthesis recipes more effectively. Moreover, during retrieval, we consider the thermodynamic relationship between target material and precursors, which is essential domain expertise in identifying the most probable precursor set among various options. Extensive experiments demonstrate the superiority of Retrieval-Retro in retrosynthesis planning, especially in discovering novel synthesis recipes, which is crucial for materials discovery. The source code for Retrieval-Retro is available at https://github.com/HeewoongNoh/Retrieval-Retro.
Protein-Mamba: Biological Mamba Models for Protein Function Prediction
Sep 22, 2024



Protein function prediction is a pivotal task in drug discovery, significantly impacting the development of effective and safe therapeutics. Traditional machine learning models often struggle with the complexity and variability inherent in predicting protein functions, necessitating more sophisticated approaches. In this work, we introduce Protein-Mamba, a novel two-stage model that leverages both self-supervised learning and fine-tuning to improve protein function prediction. The pre-training stage allows the model to capture general chemical structures and relationships from large, unlabeled datasets, while the fine-tuning stage refines these insights using specific labeled datasets, resulting in superior prediction performance. Our extensive experiments demonstrate that Protein-Mamba achieves competitive performance, compared with a couple of state-of-the-art methods across a range of protein function datasets. This model's ability to effectively utilize both unlabeled and labeled data highlights the potential of self-supervised learning in advancing protein function prediction and offers a promising direction for future research in drug discovery.
Debiased Graph Poisoning Attack via Contrastive Surrogate Objective
Jul 27, 2024



Graph neural networks (GNN) are vulnerable to adversarial attacks, which aim to degrade the performance of GNNs through imperceptible changes on the graph. However, we find that in fact the prevalent meta-gradient-based attacks, which utilizes the gradient of the loss w.r.t the adjacency matrix, are biased towards training nodes. That is, their meta-gradient is determined by a training procedure of the surrogate model, which is solely trained on the training nodes. This bias manifests as an uneven perturbation, connecting two nodes when at least one of them is a labeled node, i.e., training node, while it is unlikely to connect two unlabeled nodes. However, these biased attack approaches are sub-optimal as they do not consider flipping edges between two unlabeled nodes at all. This means that they miss the potential attacked edges between unlabeled nodes that significantly alter the representation of a node. In this paper, we investigate the meta-gradients to uncover the root cause of the uneven perturbations of existing attacks. Based on our analysis, we propose a Meta-gradient-based attack method using contrastive surrogate objective (Metacon), which alleviates the bias in meta-gradient using a new surrogate loss. We conduct extensive experiments to show that Metacon outperforms existing meta gradient-based attack methods through benchmark datasets, while showing that alleviating the bias towards training nodes is effective in attacking the graph structure.
Molecule Language Model with Augmented Pairs and Expertise Transfer
Jul 12, 2024Understanding the molecules and their textual descriptions via molecule language models (MoLM) recently got a surge of interest among researchers. However, unique challenges exist in the field of MoLM due to 1) a limited amount of molecule-text paired data and 2) missing expertise that occurred due to the specialized areas of focus among the experts. To this end, we propose AMOLE, which 1) augments molecule-text pairs with structural similarity preserving loss, and 2) transfers the expertise between the molecules. Extensive experiments on various downstream tasks demonstrate the superiority of AMOLE in comprehending molecules and their descriptions, highlighting its potential for application in real-world drug discovery.