 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMSP-LLM: A Unified Large Language Model Framework for Complete Material Synthesis Planning
Feb 07, 2026Material synthesis planning (MSP) remains a fundamental and underexplored bottleneck in AI-driven materials discovery, as it requires not only identifying suitable precursor materials but also designing coherent sequences of synthesis operations to realize a target material. Although several AI-based approaches have been proposed to address isolated subtasks of MSP, a unified methodology for solving the entire MSP task has yet to be established. We propose MSP-LLM, a unified LLM-based framework that formulates MSP as a structured process composed of two constituent subproblems: precursor prediction (PP) and synthesis operation prediction (SOP). Our approach introduces a discrete material class as an intermediate decision variable that organizes both tasks into a chemically consistent decision chain. For OP, we further incorporate hierarchical precursor types as synthesis-relevant inductive biases and employ an explicit conditioning strategy that preserves precursor-related information in the autoregressive decoding state. Extensive experiments show that MSP-LLM consistently outperforms existing methods on both PP and SOP, as well as on the complete MSP task, demonstrating an effective and scalable framework for MSP that can accelerate real-world materials discovery.
Electron-Informed Coarse-Graining Molecular Representation Learning for Real-World Molecular Physics
Feb 06, 2026Various representation learning methods for molecular structures have been devised to accelerate data-driven chemistry. However, the representation capabilities of existing methods are essentially limited to atom-level information, which is not sufficient to describe real-world molecular physics. Although electron-level information can provide fundamental knowledge about chemical compounds beyond the atom-level information, obtaining the electron-level information in real-world molecules is computationally impractical and sometimes infeasible. We propose a method for learning electron-informed molecular representations without additional computation costs by transferring readily accessible electron-level information about small molecules to large molecules of our interest. The proposed method achieved state-of-the-art prediction accuracy on extensive benchmark datasets containing experimentally observed molecular physics. The source code for HEDMoL is available at https://github.com/ngs00/HEDMoL.
3D Interaction Geometric Pre-training for Molecular Relational Learning
Dec 04, 2024Molecular Relational Learning (MRL) is a rapidly growing field that focuses on understanding the interaction dynamics between molecules, which is crucial for applications ranging from catalyst engineering to drug discovery. Despite recent progress, earlier MRL approaches are limited to using only the 2D topological structure of molecules, as obtaining the 3D interaction geometry remains prohibitively expensive. This paper introduces a novel 3D geometric pre-training strategy for MRL (3DMRL) that incorporates a 3D virtual interaction environment, overcoming the limitations of costly traditional quantum mechanical calculation methods. With the constructed 3D virtual interaction environment, 3DMRL trains 2D MRL model to learn the overall 3D geometric information of molecular interaction through contrastive learning. Moreover, fine-grained interaction between molecules is learned through force prediction loss, which is crucial in understanding the wide range of molecular interaction processes. Extensive experiments on various tasks using real-world datasets, including out-of-distribution and extrapolation scenarios, demonstrate the effectiveness of 3DMRL, showing up to a 24.93\% improvement in performance across 40 tasks.
Retrieval-Retro: Retrieval-based Inorganic Retrosynthesis with Expert Knowledge
Oct 28, 2024
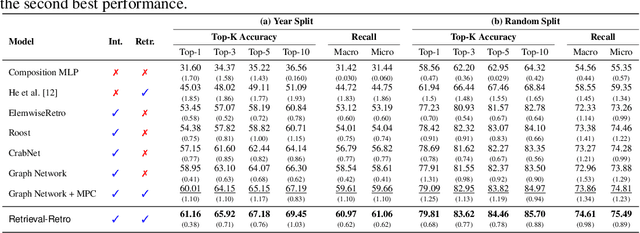
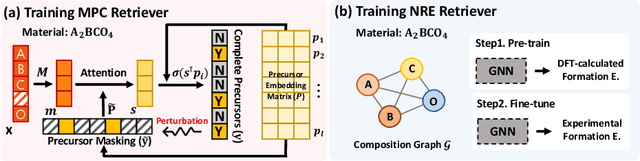
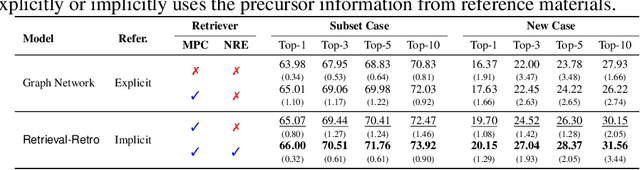
While inorganic retrosynthesis planning is essential in the field of chemical science, the application of machine learning in this area has been notably less explored compared to organic retrosynthesis planning. In this paper, we propose Retrieval-Retro for inorganic retrosynthesis planning, which implicitly extracts the precursor information of reference materials that are retrieved from the knowledge base regarding domain expertise in the field. Specifically, instead of directly employing the precursor information of reference materials, we propose implicitly extracting it with various attention layers, which enables the model to learn novel synthesis recipes more effectively. Moreover, during retrieval, we consider the thermodynamic relationship between target material and precursors, which is essential domain expertise in identifying the most probable precursor set among various options. Extensive experiments demonstrate the superiority of Retrieval-Retro in retrosynthesis planning, especially in discovering novel synthesis recipes, which is crucial for materials discovery. The source code for Retrieval-Retro is available at https://github.com/HeewoongNoh/Retrieval-Retro.
Shift-Robust Molecular Relational Learning with Causal Substructure
May 29, 2023Recently, molecular relational learning, whose goal is to predict the interaction behavior between molecular pairs, got a surge of interest in molecular sciences due to its wide range of applications. In this work, we propose CMRL that is robust to the distributional shift in molecular relational learning by detecting the core substructure that is causally related to chemical reactions. To do so, we first assume a causal relationship based on the domain knowledge of molecular sciences and construct a structural causal model (SCM) that reveals the relationship between variables. Based on the SCM, we introduce a novel conditional intervention framework whose intervention is conditioned on the paired molecule. With the conditional intervention framework, our model successfully learns from the causal substructure and alleviates the confounding effect of shortcut substructures that are spuriously correlated to chemical reactions. Extensive experiments on various tasks with real-world and synthetic datasets demonstrate the superiority of CMRL over state-of-the-art baseline models. Our code is available at https://github.com/Namkyeong/CMRL.
Conditional Graph Information Bottleneck for Molecular Relational Learning
Apr 29, 2023



Molecular relational learning, whose goal is to learn the interaction behavior between molecular pairs, got a surge of interest in molecular sciences due to its wide range of applications. Recently, graph neural networks have recently shown great success in molecular relational learning by modeling a molecule as a graph structure, and considering atom-level interactions between two molecules. Despite their success, existing molecular relational learning methods tend to overlook the nature of chemistry, i.e., a chemical compound is composed of multiple substructures such as functional groups that cause distinctive chemical reactions. In this work, we propose a novel relational learning framework, called CGIB, that predicts the interaction behavior between a pair of graphs by detecting core subgraphs therein. The main idea is, given a pair of graphs, to find a subgraph from a graph that contains the minimal sufficient information regarding the task at hand conditioned on the paired graph based on the principle of conditional graph information bottleneck. We argue that our proposed method mimics the nature of chemical reactions, i.e., the core substructure of a molecule varies depending on which other molecule it interacts with. Extensive experiments on various tasks with real-world datasets demonstrate the superiority of CGIB over state-of-the-art baselines. Our code is available at https://github.com/Namkyeong/CGIB.
Predicting Density of States via Multi-modal Transformer
Mar 13, 2023The density of states (DOS) is a spectral property of materials, which provides fundamental insights on various characteristics of materials. In this paper, we propose a model to predict the DOS by reflecting the nature of DOS: DOS determines the general distribution of states as a function of energy. Specifically, we integrate the heterogeneous information obtained from the crystal structure and the energies via multi-modal transformer, thereby modeling the complex relationships between the atoms in the crystal structure, and various energy levels. Extensive experiments on two types of DOS, i.e., Phonon DOS and Electron DOS, with various real-world scenarios demonstrate the superiority of DOSTransformer. The source code for DOSTransformer is available at https://github.com/HeewoongNoh/DOSTransformer.