 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImage is All You Need: Towards Efficient and Effective Large Language Model-Based Recommender Systems
Mar 08, 2025Large Language Models (LLMs) have recently emerged as a powerful backbone for recommender systems. Existing LLM-based recommender systems take two different approaches for representing items in natural language, i.e., Attribute-based Representation and Description-based Representation. In this work, we aim to address the trade-off between efficiency and effectiveness that these two approaches encounter, when representing items consumed by users. Based on our interesting observation that there is a significant information overlap between images and descriptions associated with items, we propose a novel method, Image is all you need for LLM-based Recommender system (I-LLMRec). Our main idea is to leverage images as an alternative to lengthy textual descriptions for representing items, aiming at reducing token usage while preserving the rich semantic information of item descriptions. Through extensive experiments, we demonstrate that I-LLMRec outperforms existing methods in both efficiency and effectiveness by leveraging images. Moreover, a further appeal of I-LLMRec is its ability to reduce sensitivity to noise in descriptions, leading to more robust recommendations.
3D Interaction Geometric Pre-training for Molecular Relational Learning
Dec 04, 2024Molecular Relational Learning (MRL) is a rapidly growing field that focuses on understanding the interaction dynamics between molecules, which is crucial for applications ranging from catalyst engineering to drug discovery. Despite recent progress, earlier MRL approaches are limited to using only the 2D topological structure of molecules, as obtaining the 3D interaction geometry remains prohibitively expensive. This paper introduces a novel 3D geometric pre-training strategy for MRL (3DMRL) that incorporates a 3D virtual interaction environment, overcoming the limitations of costly traditional quantum mechanical calculation methods. With the constructed 3D virtual interaction environment, 3DMRL trains 2D MRL model to learn the overall 3D geometric information of molecular interaction through contrastive learning. Moreover, fine-grained interaction between molecules is learned through force prediction loss, which is crucial in understanding the wide range of molecular interaction processes. Extensive experiments on various tasks using real-world datasets, including out-of-distribution and extrapolation scenarios, demonstrate the effectiveness of 3DMRL, showing up to a 24.93\% improvement in performance across 40 tasks.
Retrieval-Retro: Retrieval-based Inorganic Retrosynthesis with Expert Knowledge
Oct 28, 2024
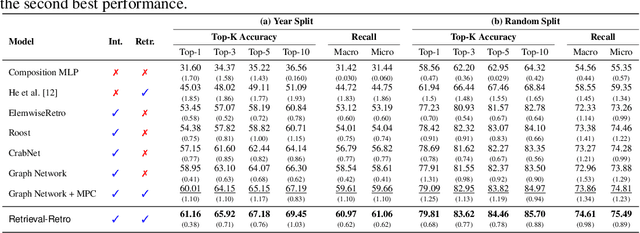
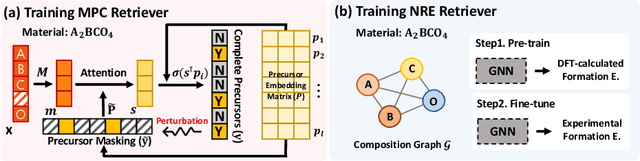
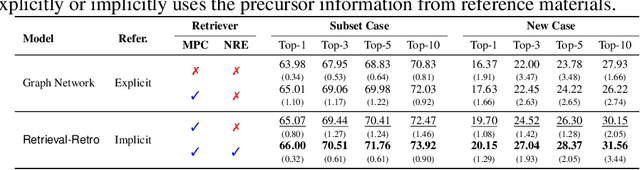
While inorganic retrosynthesis planning is essential in the field of chemical science, the application of machine learning in this area has been notably less explored compared to organic retrosynthesis planning. In this paper, we propose Retrieval-Retro for inorganic retrosynthesis planning, which implicitly extracts the precursor information of reference materials that are retrieved from the knowledge base regarding domain expertise in the field. Specifically, instead of directly employing the precursor information of reference materials, we propose implicitly extracting it with various attention layers, which enables the model to learn novel synthesis recipes more effectively. Moreover, during retrieval, we consider the thermodynamic relationship between target material and precursors, which is essential domain expertise in identifying the most probable precursor set among various options. Extensive experiments demonstrate the superiority of Retrieval-Retro in retrosynthesis planning, especially in discovering novel synthesis recipes, which is crucial for materials discovery. The source code for Retrieval-Retro is available at https://github.com/HeewoongNoh/Retrieval-Retro.
Predicting Density of States via Multi-modal Transformer
Mar 13, 2023The density of states (DOS) is a spectral property of materials, which provides fundamental insights on various characteristics of materials. In this paper, we propose a model to predict the DOS by reflecting the nature of DOS: DOS determines the general distribution of states as a function of energy. Specifically, we integrate the heterogeneous information obtained from the crystal structure and the energies via multi-modal transformer, thereby modeling the complex relationships between the atoms in the crystal structure, and various energy levels. Extensive experiments on two types of DOS, i.e., Phonon DOS and Electron DOS, with various real-world scenarios demonstrate the superiority of DOSTransformer. The source code for DOSTransformer is available at https://github.com/HeewoongNoh/DOSTransformer.