 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtein-Mamba: Biological Mamba Models for Protein Function Prediction
Sep 22, 2024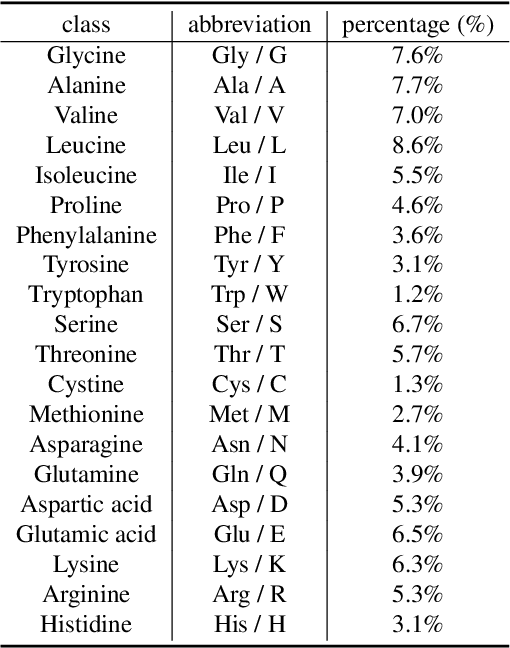



Protein function prediction is a pivotal task in drug discovery, significantly impacting the development of effective and safe therapeutics. Traditional machine learning models often struggle with the complexity and variability inherent in predicting protein functions, necessitating more sophisticated approaches. In this work, we introduce Protein-Mamba, a novel two-stage model that leverages both self-supervised learning and fine-tuning to improve protein function prediction. The pre-training stage allows the model to capture general chemical structures and relationships from large, unlabeled datasets, while the fine-tuning stage refines these insights using specific labeled datasets, resulting in superior prediction performance. Our extensive experiments demonstrate that Protein-Mamba achieves competitive performance, compared with a couple of state-of-the-art methods across a range of protein function datasets. This model's ability to effectively utilize both unlabeled and labeled data highlights the potential of self-supervised learning in advancing protein function prediction and offers a promising direction for future research in drug discovery.
SMILES-Mamba: Chemical Mamba Foundation Models for Drug ADMET Prediction
Aug 11, 2024
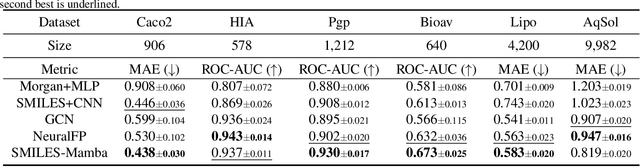


In drug discovery, predicting the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of small-molecule drugs is critical for ensuring safety and efficacy. However, the process of accurately predicting these properties is often resource-intensive and requires extensive experimental data. To address this challenge, we propose SMILES-Mamba, a two-stage model that leverages both unlabeled and labeled data through a combination of self-supervised pretraining and fine-tuning strategies. The model first pre-trains on a large corpus of unlabeled SMILES strings to capture the underlying chemical structure and relationships, before being fine-tuned on smaller, labeled datasets specific to ADMET tasks. Our results demonstrate that SMILES-Mamba exhibits competitive performance across 22 ADMET datasets, achieving the highest score in 14 tasks, highlighting the potential of self-supervised learning in improving molecular property prediction. This approach not only enhances prediction accuracy but also reduces the dependence on large, labeled datasets, offering a promising direction for future research in drug discovery.
AntibodyFlow: Normalizing Flow Model for Designing Antibody Complementarity-Determining Regions
Jun 19, 2024



Therapeutic antibodies have been extensively studied in drug discovery and development in the past decades. Antibodies are specialized protective proteins that bind to antigens in a lock-to-key manner. The binding strength/affinity between an antibody and a specific antigen is heavily determined by the complementarity-determining regions (CDRs) on the antibodies. Existing machine learning methods cast in silico development of CDRs as either sequence or 3D graph (with a single chain) generation tasks and have achieved initial success. However, with CDR loops having specific geometry shapes, learning the 3D geometric structures of CDRs remains a challenge. To address this issue, we propose AntibodyFlow, a 3D flow model to design antibody CDR loops. Specifically, AntibodyFlow first constructs the distance matrix, then predicts amino acids conditioned on the distance matrix. Also, AntibodyFlow conducts constraint learning and constrained generation to ensure valid 3D structures. Experimental results indicate that AntibodyFlow outperforms the best baseline consistently with up to 16.0% relative improvement in validity rate and 24.3% relative reduction in geometric graph level error (root mean square deviation, RMSD).