 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSMILES-Mamba: Chemical Mamba Foundation Models for Drug ADMET Prediction
Paper and Code
Aug 11, 2024
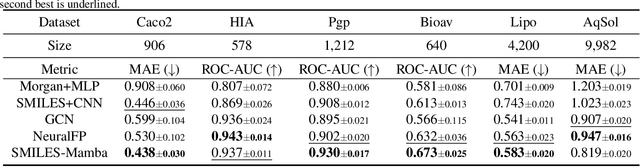


In drug discovery, predicting the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of small-molecule drugs is critical for ensuring safety and efficacy. However, the process of accurately predicting these properties is often resource-intensive and requires extensive experimental data. To address this challenge, we propose SMILES-Mamba, a two-stage model that leverages both unlabeled and labeled data through a combination of self-supervised pretraining and fine-tuning strategies. The model first pre-trains on a large corpus of unlabeled SMILES strings to capture the underlying chemical structure and relationships, before being fine-tuned on smaller, labeled datasets specific to ADMET tasks. Our results demonstrate that SMILES-Mamba exhibits competitive performance across 22 ADMET datasets, achieving the highest score in 14 tasks, highlighting the potential of self-supervised learning in improving molecular property prediction. This approach not only enhances prediction accuracy but also reduces the dependence on large, labeled datasets, offering a promising direction for future research in drug discovery.