 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutoEval Done Right: Using Synthetic Data for Model Evaluation
Mar 09, 2024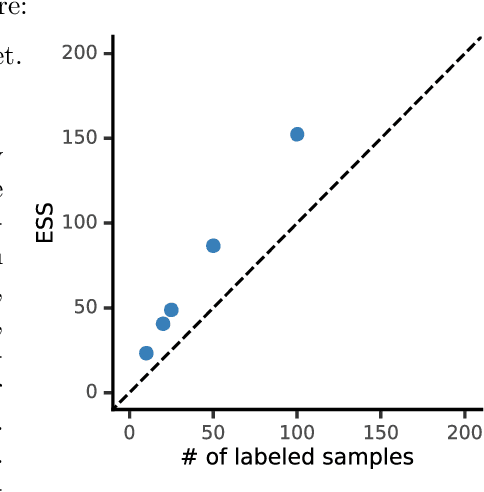
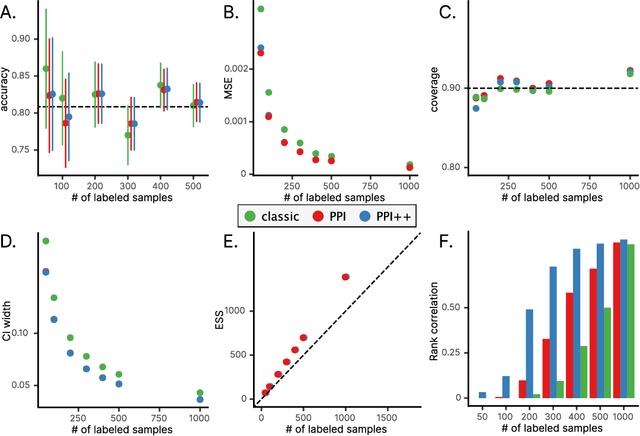
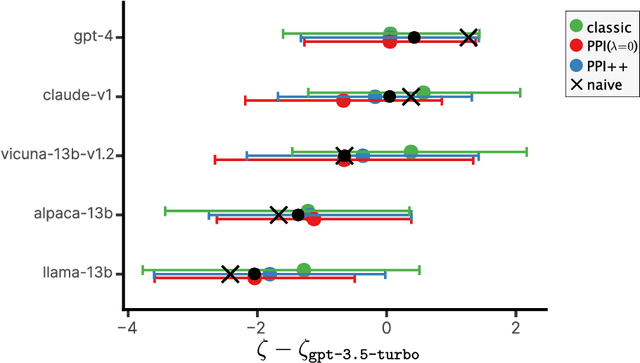
The evaluation of machine learning models using human-labeled validation data can be expensive and time-consuming. AI-labeled synthetic data can be used to decrease the number of human annotations required for this purpose in a process called autoevaluation. We suggest efficient and statistically principled algorithms for this purpose that improve sample efficiency while remaining unbiased. These algorithms increase the effective human-labeled sample size by up to 50% on experiments with GPT-4.
Decision-Making with Auto-Encoding Variational Bayes
Feb 17, 2020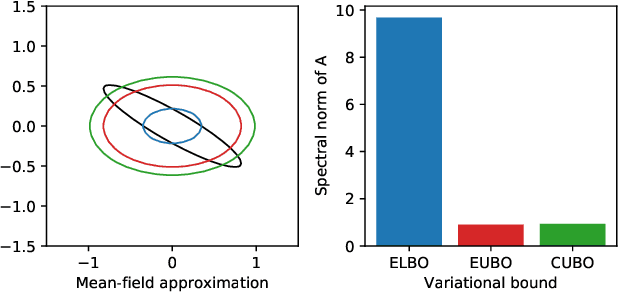

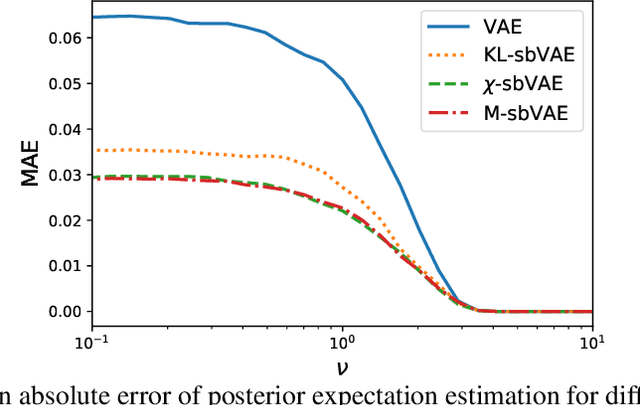

To make decisions based on a model fit by Auto-Encoding Variational Bayes (AEVB), practitioners typically use importance sampling to estimate a functional of the posterior distribution. The variational distribution found by AEVB serves as the proposal distribution for importance sampling. However, this proposal distribution may give unreliable (high variance) importance sampling estimates, thus leading to poor decisions. We explore how changing the objective function for learning the variational distribution, while continuing to learn the generative model based on the ELBO, affects the quality of downstream decisions. For a particular model, we characterize the error of importance sampling as a function of posterior variance and show that proposal distributions learned with evidence upper bounds are better. Motivated by these theoretical results, we propose a novel variant of the VAE. In addition to experimenting with MNIST, we present a full-fledged application of the proposed method to single-cell RNA sequencing. In this challenging instance of multiple hypothesis testing, the proposed method surpasses the current state of the art.
A joint model of unpaired data from scRNA-seq and spatial transcriptomics for imputing missing gene expression measurements
May 06, 2019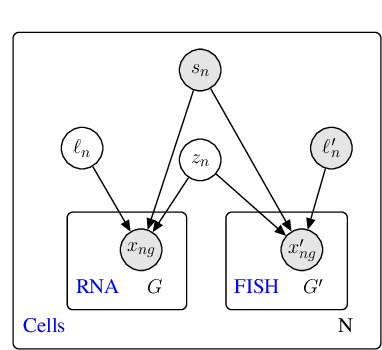
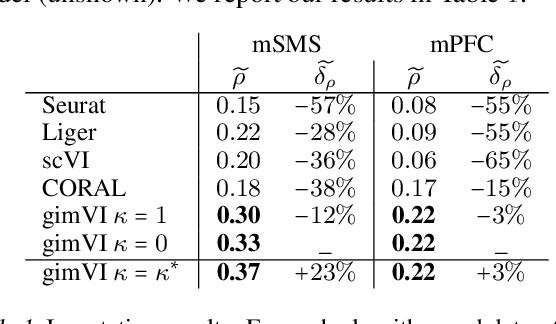
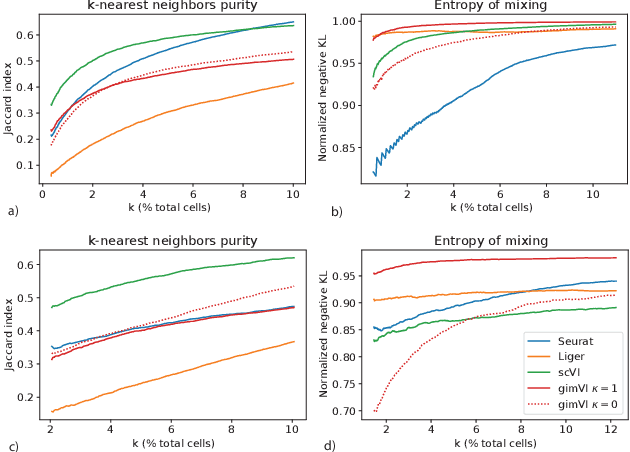
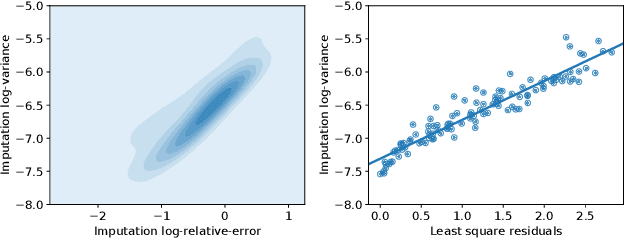
Spatial studies of transcriptome provide biologists with gene expression maps of heterogeneous and complex tissues. However, most experimental protocols for spatial transcriptomics suffer from the need to select beforehand a small fraction of genes to be quantified over the entire transcriptome. Standard single-cell RNA sequencing (scRNA-seq) is more prevalent, easier to implement and can in principle capture any gene but cannot recover the spatial location of the cells. In this manuscript, we focus on the problem of imputation of missing genes in spatial transcriptomic data based on (unpaired) standard scRNA-seq data from the same biological tissue. Building upon domain adaptation work, we propose gimVI, a deep generative model for the integration of spatial transcriptomic data and scRNA-seq data that can be used to impute missing genes. After describing our generative model and an inference procedure for it, we compare gimVI to alternative methods from computational biology or domain adaptation on real datasets and outperform Seurat Anchors, Liger and CORAL to impute held-out genes.
Information Constraints on Auto-Encoding Variational Bayes
Oct 15, 2018
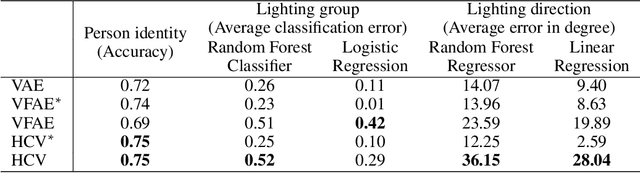


Parameterizing the approximate posterior of a generative model with neural networks has become a common theme in recent machine learning research. While providing appealing flexibility, this approach makes it difficult to impose or assess structural constraints such as conditional independence. We propose a framework for learning representations that relies on Auto-Encoding Variational Bayes and whose search space is constrained via kernel-based measures of independence. In particular, our method employs the $d$-variable Hilbert-Schmidt Independence Criterion (dHSIC) to enforce independence between the latent representations and arbitrary nuisance factors. We show how to apply this method to a range of problems, including the problems of learning invariant representations and the learning of interpretable representations. We also present a full-fledged application to single-cell RNA sequencing (scRNA-seq). In this setting the biological signal is mixed in complex ways with sequencing errors and sampling effects. We show that our method out-performs the state-of-the-art in this domain.
A Deep Generative Model for Semi-Supervised Classification with Noisy Labels
Sep 16, 2018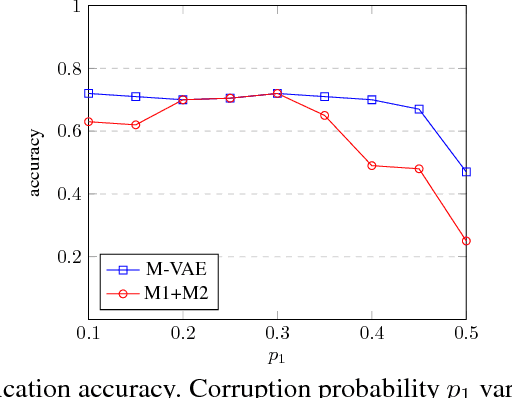
Class labels are often imperfectly observed, due to mistakes and to genuine ambiguity among classes. We propose a new semi-supervised deep generative model that explicitly models noisy labels, called the Mislabeled VAE (M-VAE). The M-VAE can perform better than existing deep generative models which do not account for label noise. Additionally, the derivation of M-VAE gives new theoretical insights into the popular M1+M2 semi-supervised model.
A deep generative model for gene expression profiles from single-cell RNA sequencing
Jan 16, 2018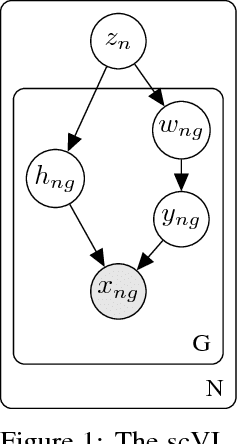



We propose a probabilistic model for interpreting gene expression levels that are observed through single-cell RNA sequencing. In the model, each cell has a low-dimensional latent representation. Additional latent variables account for technical effects that may erroneously set some observations of gene expression levels to zero. Conditional distributions are specified by neural networks, giving the proposed model enough flexibility to fit the data well. We use variational inference and stochastic optimization to approximate the posterior distribution. The inference procedure scales to over one million cells, whereas competing algorithms do not. Even for smaller datasets, for several tasks, the proposed procedure outperforms state-of-the-art methods like ZIFA and ZINB-WaVE. We also extend our framework to account for batch effects and other confounding factors, and propose a Bayesian hypothesis test for differential expression that outperforms DESeq2.
A deep generative model for single-cell RNA sequencing with application to detecting differentially expressed genes
Oct 17, 2017

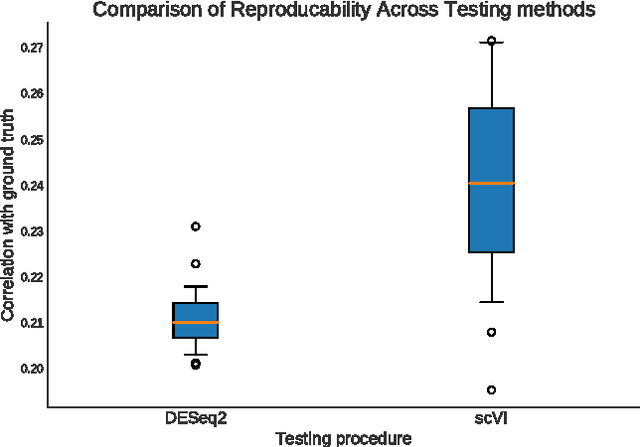

We propose a probabilistic model for interpreting gene expression levels that are observed through single-cell RNA sequencing. In the model, each cell has a low-dimensional latent representation. Additional latent variables account for technical effects that may erroneously set some observations of gene expression levels to zero. Conditional distributions are specified by neural networks, giving the proposed model enough flexibility to fit the data well. We use variational inference and stochastic optimization to approximate the posterior distribution. The inference procedure scales to over one million cells, whereas competing algorithms do not. Even for smaller datasets, for several tasks, the proposed procedure outperforms state-of-the-art methods like ZIFA and ZINB-WaVE. We also extend our framework to take into account batch effects and other confounding factors and propose a natural Bayesian hypothesis framework for differential expression that outperforms tradition DESeq2.