 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA deep generative model for gene expression profiles from single-cell RNA sequencing
Jan 16, 2018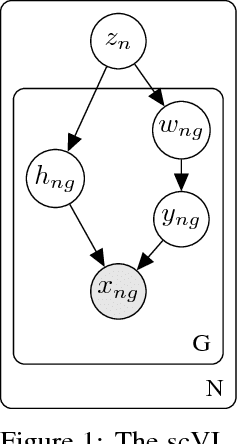



We propose a probabilistic model for interpreting gene expression levels that are observed through single-cell RNA sequencing. In the model, each cell has a low-dimensional latent representation. Additional latent variables account for technical effects that may erroneously set some observations of gene expression levels to zero. Conditional distributions are specified by neural networks, giving the proposed model enough flexibility to fit the data well. We use variational inference and stochastic optimization to approximate the posterior distribution. The inference procedure scales to over one million cells, whereas competing algorithms do not. Even for smaller datasets, for several tasks, the proposed procedure outperforms state-of-the-art methods like ZIFA and ZINB-WaVE. We also extend our framework to account for batch effects and other confounding factors, and propose a Bayesian hypothesis test for differential expression that outperforms DESeq2.
A deep generative model for single-cell RNA sequencing with application to detecting differentially expressed genes
Oct 17, 2017

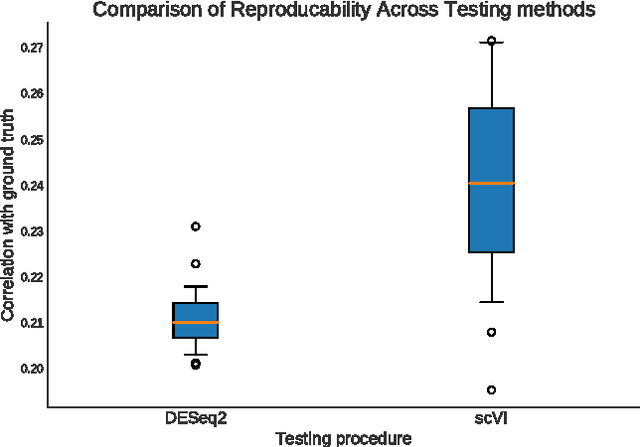

We propose a probabilistic model for interpreting gene expression levels that are observed through single-cell RNA sequencing. In the model, each cell has a low-dimensional latent representation. Additional latent variables account for technical effects that may erroneously set some observations of gene expression levels to zero. Conditional distributions are specified by neural networks, giving the proposed model enough flexibility to fit the data well. We use variational inference and stochastic optimization to approximate the posterior distribution. The inference procedure scales to over one million cells, whereas competing algorithms do not. Even for smaller datasets, for several tasks, the proposed procedure outperforms state-of-the-art methods like ZIFA and ZINB-WaVE. We also extend our framework to take into account batch effects and other confounding factors and propose a natural Bayesian hypothesis framework for differential expression that outperforms tradition DESeq2.