 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScaffold-constrained molecular generation
Oct 05, 2020
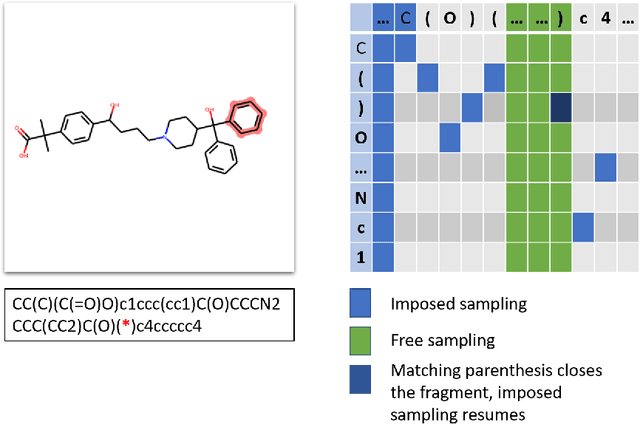
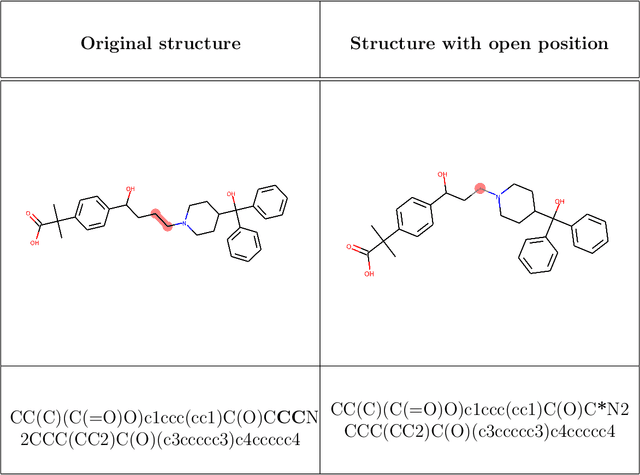

One of the major applications of generative models for drug Discovery targets the lead-optimization phase. During the optimization of a lead series, it is common to have scaffold constraints imposed on the structure of the molecules designed. Without enforcing such constraints, the probability of generating molecules with the required scaffold is extremely low and hinders the practicality of generative models for de-novo drug design. To tackle this issue, we introduce a new algorithm to perform scaffold-constrained in-silico molecular design. We build on the well-known SMILES-based Recurrent Neural Network (RNN) generative model, with a modified sampling procedure to achieve scaffold-constrained generation. We directly benefit from the associated reinforcement Learning methods, allowing to design molecules optimized for different properties while exploring only the relevant chemical space. We showcase the method's ability to perform scaffold-constrained generation on various tasks: designing novel molecules around scaffolds extracted from SureChEMBL chemical series, generating novel active molecules on the Dopamine Receptor D2 (DRD2) target, and, finally, designing predicted actives on the MMP-12 series, an industrial lead-optimization project.
A joint model of unpaired data from scRNA-seq and spatial transcriptomics for imputing missing gene expression measurements
May 06, 2019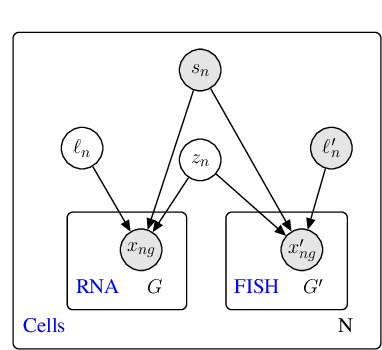
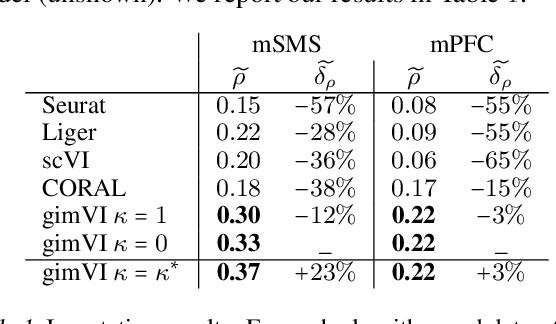
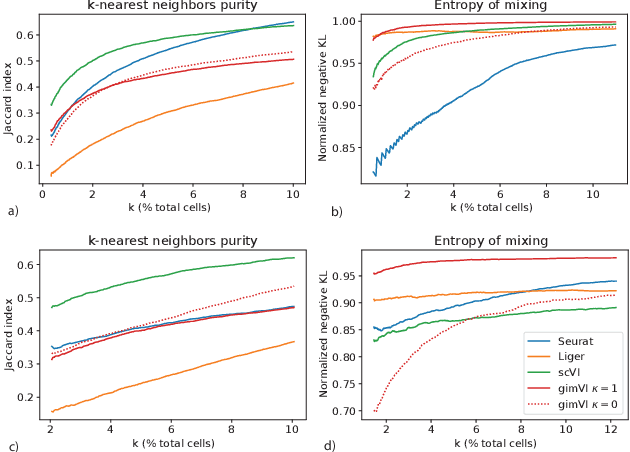
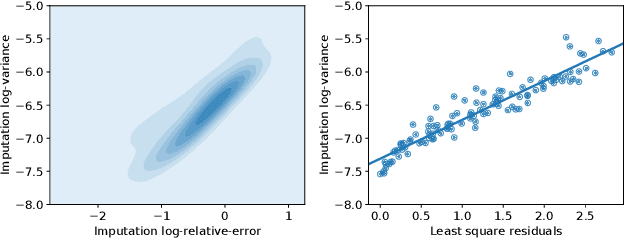
Spatial studies of transcriptome provide biologists with gene expression maps of heterogeneous and complex tissues. However, most experimental protocols for spatial transcriptomics suffer from the need to select beforehand a small fraction of genes to be quantified over the entire transcriptome. Standard single-cell RNA sequencing (scRNA-seq) is more prevalent, easier to implement and can in principle capture any gene but cannot recover the spatial location of the cells. In this manuscript, we focus on the problem of imputation of missing genes in spatial transcriptomic data based on (unpaired) standard scRNA-seq data from the same biological tissue. Building upon domain adaptation work, we propose gimVI, a deep generative model for the integration of spatial transcriptomic data and scRNA-seq data that can be used to impute missing genes. After describing our generative model and an inference procedure for it, we compare gimVI to alternative methods from computational biology or domain adaptation on real datasets and outperform Seurat Anchors, Liger and CORAL to impute held-out genes.
A Deep Generative Model for Semi-Supervised Classification with Noisy Labels
Sep 16, 2018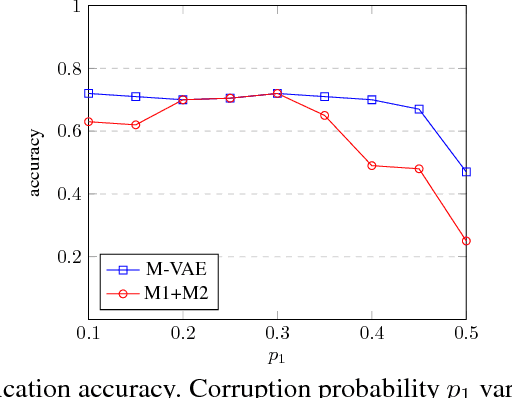
Class labels are often imperfectly observed, due to mistakes and to genuine ambiguity among classes. We propose a new semi-supervised deep generative model that explicitly models noisy labels, called the Mislabeled VAE (M-VAE). The M-VAE can perform better than existing deep generative models which do not account for label noise. Additionally, the derivation of M-VAE gives new theoretical insights into the popular M1+M2 semi-supervised model.