 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIceCache: Memory-efficient KV-cache Management for Long-Sequence LLMs
Apr 12, 2026Key-Value (KV) cache plays a crucial role in accelerating inference in large language models (LLMs) by storing intermediate attention states and avoiding redundant computation during autoregressive generation. However, its memory footprint scales linearly with sequence length, often leading to severe memory bottlenecks on resource-constrained hardware. Prior work has explored offloading KV cache to the CPU while retaining only a subset on the GPU, but these approaches often rely on imprecise token selection and suffer performance degradation in long-generation tasks such as chain-of-thought reasoning. In this paper, we propose a novel KV cache management strategy, IceCache, which integrates semantic token clustering with PagedAttention. By organizing semantically related tokens into contiguous memory regions managed by a hierarchical, dynamically updatable data structure, our method enables more efficient token selection and better utilization of memory bandwidth during CPU-GPU transfers. Experimental results on LongBench show that, with a 256-token budget, IceCache maintains 99% of the original accuracy achieved by the full KV cache model. Moreover, compared to other offloading-based methods, IceCache attains competitive or even superior latency and accuracy while using only 25% of the KV cache token budget, demonstrating its effectiveness in long-sequence scenarios. The code is available on our project website at https://yuzhenmao.github.io/IceCache/.
PAS: Estimating the target accuracy before domain adaptation
Apr 10, 2026The goal of domain adaptation is to make predictions for unlabeled samples from a target domain with the help of labeled samples from a different but related source domain. The performance of domain adaptation methods is highly influenced by the choice of source domain and pre-trained feature extractor. However, the selection of source data and pre-trained model is not trivial due to the absence of a labeled validation set for the target domain and the large number of available pre-trained models. In this work, we propose PAS, a novel score designed to estimate the transferability of a source domain set and a pre-trained feature extractor to a target classification task before actually performing domain adaptation. PAS leverages the generalization power of pre-trained models and assesses source-target compatibility based on the pre-trained feature embeddings. We integrate PAS into a framework that indicates the most relevant pre-trained model and source domain among multiple candidates, thus improving target accuracy while reducing the computational overhead. Extensive experiments on image classification benchmarks demonstrate that PAS correlates strongly with actual target accuracy and consistently guides the selection of the best-performing pre-trained model and source domain for adaptation.
Compositional Flows for 3D Molecule and Synthesis Pathway Co-design
Apr 10, 2025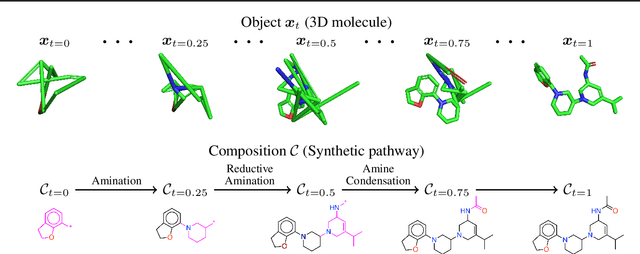
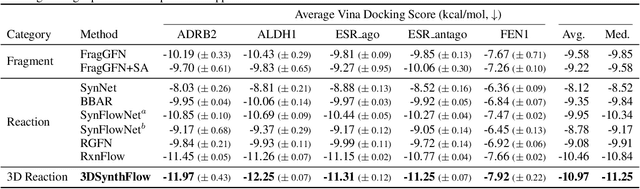
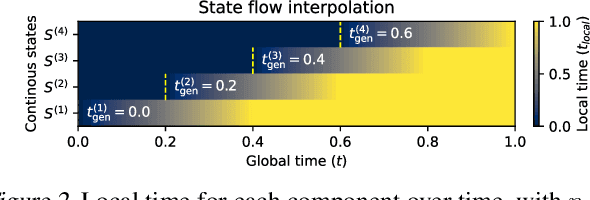
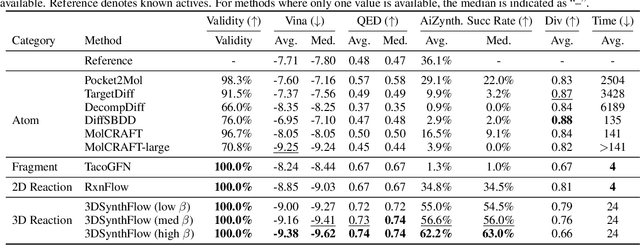
Many generative applications, such as synthesis-based 3D molecular design, involve constructing compositional objects with continuous features. Here, we introduce Compositional Generative Flows (CGFlow), a novel framework that extends flow matching to generate objects in compositional steps while modeling continuous states. Our key insight is that modeling compositional state transitions can be formulated as a straightforward extension of the flow matching interpolation process. We further build upon the theoretical foundations of generative flow networks (GFlowNets), enabling reward-guided sampling of compositional structures. We apply CGFlow to synthesizable drug design by jointly designing the molecule's synthetic pathway with its 3D binding pose. Our approach achieves state-of-the-art binding affinity on all 15 targets from the LIT-PCBA benchmark, and 5.8$\times$ improvement in sampling efficiency compared to 2D synthesis-based baseline. To our best knowledge, our method is also the first to achieve state of-art-performance in both Vina Dock (-9.38) and AiZynth success rate (62.2\%) on the CrossDocked benchmark.
Improving OOD Generalization of Pre-trained Encoders via Aligned Embedding-Space Ensembles
Nov 20, 2024



The quality of self-supervised pre-trained embeddings on out-of-distribution (OOD) data is poor without fine-tuning. A straightforward and simple approach to improving the generalization of pre-trained representation to OOD data is the use of deep ensembles. However, obtaining an effective ensemble in the embedding space with only unlabeled data remains an unsolved problem. We first perform a theoretical analysis that reveals the relationship between individual hyperspherical embedding spaces in an ensemble. We then design a principled method to align these embedding spaces in an unsupervised manner. Experimental results on the MNIST dataset show that our embedding-space ensemble method improves pre-trained embedding quality on in-distribution and OOD data compared to single encoders.
Causal Order Discovery based on Monotonic SCMs
Oct 24, 2024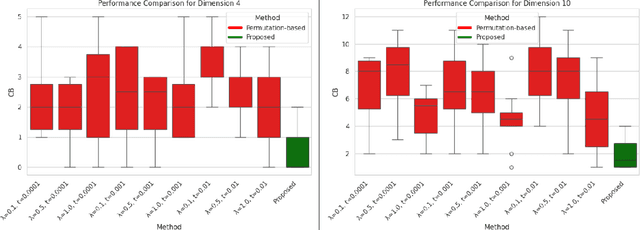
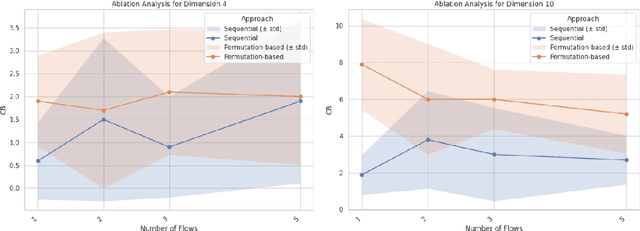

In this paper, we consider the problem of causal order discovery within the framework of monotonic Structural Causal Models (SCMs), which have gained attention for their potential to enable causal inference and causal discovery from observational data. While existing approaches either assume prior knowledge about the causal order or use complex optimization techniques to impose sparsity in the Jacobian of Triangular Monotonic Increasing maps, our work introduces a novel sequential procedure that directly identifies the causal order by iteratively detecting the root variable. This method eliminates the need for sparsity assumptions and the associated optimization challenges, enabling the identification of a unique SCM without the need for multiple independence tests to break the Markov equivalence class. We demonstrate the effectiveness of our approach in sequentially finding the root variable, comparing it to methods that maximize Jacobian sparsity.
Generative Flows on Synthetic Pathway for Drug Design
Oct 06, 2024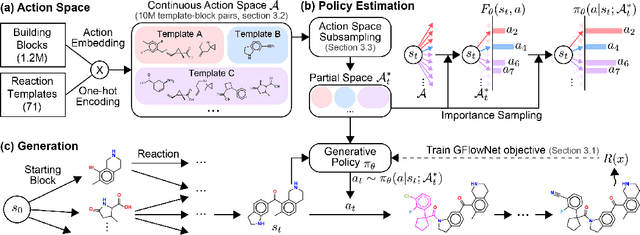
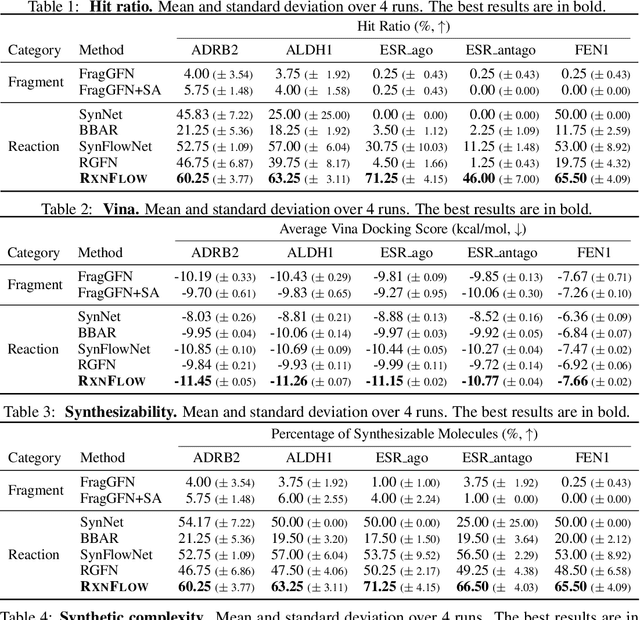
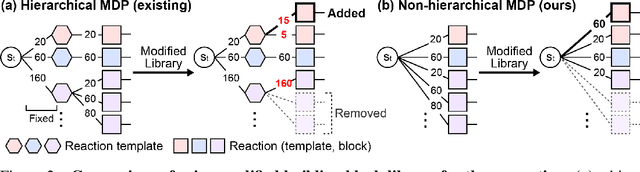
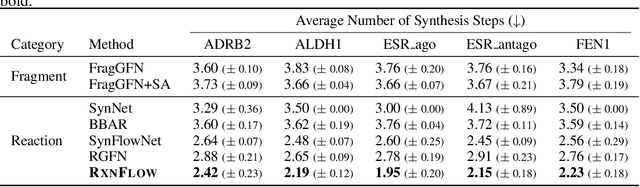
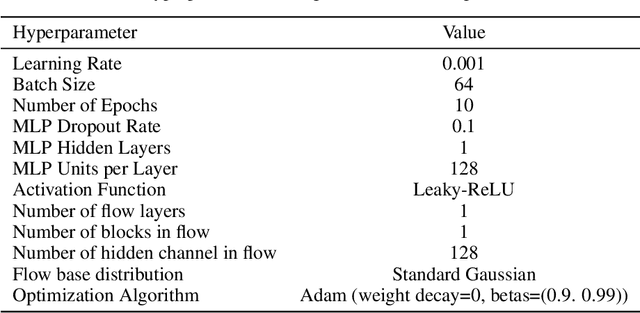
Generative models in drug discovery have recently gained attention as efficient alternatives to brute-force virtual screening. However, most existing models do not account for synthesizability, limiting their practical use in real-world scenarios. In this paper, we propose RxnFlow, which sequentially assembles molecules using predefined molecular building blocks and chemical reaction templates to constrain the synthetic chemical pathway. We then train on this sequential generating process with the objective of generative flow networks (GFlowNets) to generate both highly rewarded and diverse molecules. To mitigate the large action space of synthetic pathways in GFlowNets, we implement a novel action space subsampling method. This enables RxnFlow to learn generative flows over extensive action spaces comprising combinations of 1.2 million building blocks and 71 reaction templates without significant computational overhead. Additionally, RxnFlow can employ modified or expanded action spaces for generation without retraining, allowing for the introduction of additional objectives or the incorporation of newly discovered building blocks. We experimentally demonstrate that RxnFlow outperforms existing reaction-based and fragment-based models in pocket-specific optimization across various target pockets. Furthermore, RxnFlow achieves state-of-the-art performance on CrossDocked2020 for pocket-conditional generation, with an average Vina score of -8.85kcal/mol and 34.8% synthesizability.
UnPaSt: unsupervised patient stratification by differentially expressed biclusters in omics data
Jul 31, 2024Most complex diseases, including cancer and non-malignant diseases like asthma, have distinct molecular subtypes that require distinct clinical approaches. However, existing computational patient stratification methods have been benchmarked almost exclusively on cancer omics data and only perform well when mutually exclusive subtypes can be characterized by many biomarkers. Here, we contribute with a massive evaluation attempt, quantitatively exploring the power of 22 unsupervised patient stratification methods using both, simulated and real transcriptome data. From this experience, we developed UnPaSt (https://apps.cosy.bio/unpast/) optimizing unsupervised patient stratification, working even with only a limited number of subtype-predictive biomarkers. We evaluated all 23 methods on real-world breast cancer and asthma transcriptomics data. Although many methods reliably detected major breast cancer subtypes, only few identified Th2-high asthma, and UnPaSt significantly outperformed its closest competitors in both test datasets. Essentially, we showed that UnPaSt can detect many biologically insightful and reproducible patterns in omic datasets.
Geometric-informed GFlowNets for Structure-Based Drug Design
Jun 16, 2024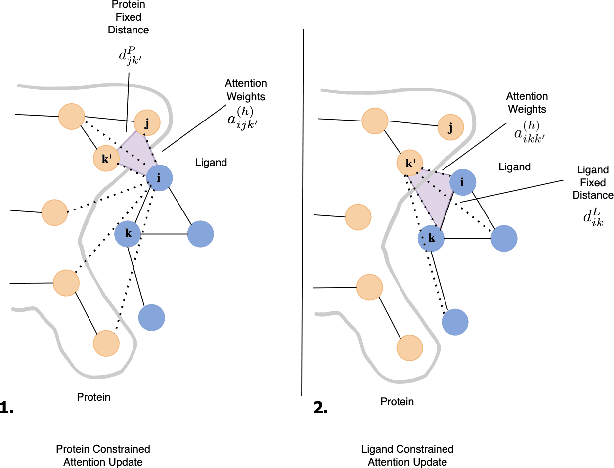


The rise of cost involved with drug discovery and current speed of which they are discover, underscore the need for more efficient structure-based drug design (SBDD) methods. We employ Generative Flow Networks (GFlowNets), to effectively explore the vast combinatorial space of drug-like molecules, which traditional virtual screening methods fail to cover. We introduce a novel modification to the GFlowNet framework by incorporating trigonometrically consistent embeddings, previously utilized in tasks involving protein conformation and protein-ligand interactions, to enhance the model's ability to generate molecules tailored to specific protein pockets. We have modified the existing protein conditioning used by GFlowNets, blending geometric information from both protein and ligand embeddings to achieve more geometrically consistent embeddings. Experiments conducted using CrossDocked2020 demonstrated an improvement in the binding affinity between generated molecules and protein pockets for both single and multi-objective tasks, compared to previous work. Additionally, we propose future work aimed at further increasing the geometric information captured in protein-ligand interactions.
IceFormer: Accelerated Inference with Long-Sequence Transformers on CPUs
May 05, 2024



One limitation of existing Transformer-based models is that they cannot handle very long sequences as input since their self-attention operations exhibit quadratic time and space complexity. This problem becomes especially acute when Transformers are deployed on hardware platforms equipped only with CPUs. To address this issue, we propose a novel method for accelerating self-attention at inference time that works with pretrained Transformer models out-of-the-box without requiring retraining. We experiment using our method to accelerate various long-sequence Transformers, including a leading LLaMA 2-based LLM, on various benchmarks and demonstrate a greater speedup of 2.73x - 7.63x while retaining 98.6% - 99.6% of the accuracy of the original pretrained models. The code is available on our project website at https://yuzhenmao.github.io/IceFormer/.
Adversarially Balanced Representation for Continuous Treatment Effect Estimation
Dec 17, 2023Individual treatment effect (ITE) estimation requires adjusting for the covariate shift between populations with different treatments, and deep representation learning has shown great promise in learning a balanced representation of covariates. However the existing methods mostly consider the scenario of binary treatments. In this paper, we consider the more practical and challenging scenario in which the treatment is a continuous variable (e.g. dosage of a medication), and we address the two main challenges of this setup. We propose the adversarial counterfactual regression network (ACFR) that adversarially minimizes the representation imbalance in terms of KL divergence, and also maintains the impact of the treatment value on the outcome prediction by leveraging an attention mechanism. Theoretically we demonstrate that ACFR objective function is grounded in an upper bound on counterfactual outcome prediction error. Our experimental evaluation on semi-synthetic datasets demonstrates the empirical superiority of ACFR over a range of state-of-the-art methods.