 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePretraining Generative Flow Networks with Inexpensive Rewards for Molecular Graph Generation
Mar 08, 2025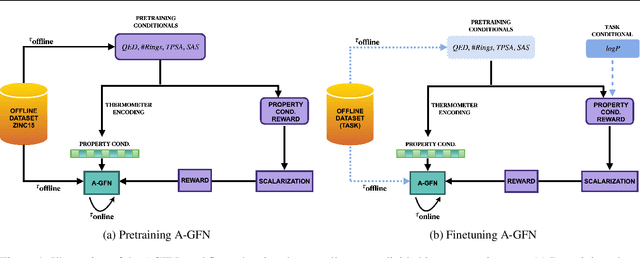



Generative Flow Networks (GFlowNets) have recently emerged as a suitable framework for generating diverse and high-quality molecular structures by learning from rewards treated as unnormalized distributions. Previous works in this framework often restrict exploration by using predefined molecular fragments as building blocks, limiting the chemical space that can be accessed. In this work, we introduce Atomic GFlowNets (A-GFNs), a foundational generative model leveraging individual atoms as building blocks to explore drug-like chemical space more comprehensively. We propose an unsupervised pre-training approach using drug-like molecule datasets, which teaches A-GFNs about inexpensive yet informative molecular descriptors such as drug-likeliness, topological polar surface area, and synthetic accessibility scores. These properties serve as proxy rewards, guiding A-GFNs towards regions of chemical space that exhibit desirable pharmacological properties. We further implement a goal-conditioned finetuning process, which adapts A-GFNs to optimize for specific target properties. In this work, we pretrain A-GFN on a subset of ZINC dataset, and by employing robust evaluation metrics we show the effectiveness of our approach when compared to other relevant baseline methods for a wide range of drug design tasks.
GFlowNet Pretraining with Inexpensive Rewards
Sep 15, 2024



Generative Flow Networks (GFlowNets), a class of generative models have recently emerged as a suitable framework for generating diverse and high-quality molecular structures by learning from unnormalized reward distributions. Previous works in this direction often restrict exploration by using predefined molecular fragments as building blocks, limiting the chemical space that can be accessed. In this work, we introduce Atomic GFlowNets (A-GFNs), a foundational generative model leveraging individual atoms as building blocks to explore drug-like chemical space more comprehensively. We propose an unsupervised pre-training approach using offline drug-like molecule datasets, which conditions A-GFNs on inexpensive yet informative molecular descriptors such as drug-likeliness, topological polar surface area, and synthetic accessibility scores. These properties serve as proxy rewards, guiding A-GFNs towards regions of chemical space that exhibit desirable pharmacological properties. We further our method by implementing a goal-conditioned fine-tuning process, which adapts A-GFNs to optimize for specific target properties. In this work, we pretrain A-GFN on the ZINC15 offline dataset and employ robust evaluation metrics to show the effectiveness of our approach when compared to other relevant baseline methods in drug design.
TacoGFN: Target Conditioned GFlowNet for Structure-Based Drug Design
Oct 05, 2023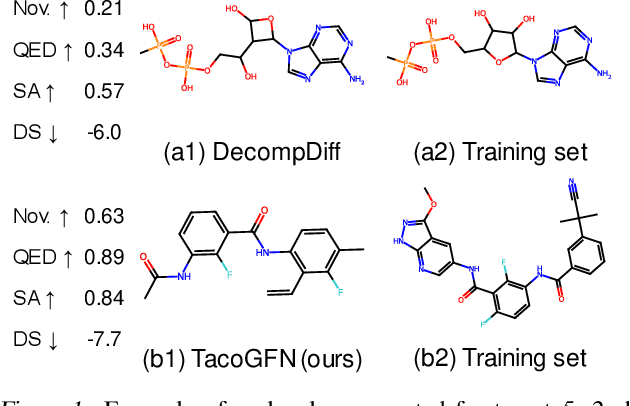



We seek to automate the generation of drug-like compounds conditioned to specific protein pocket targets. Most current methods approximate the protein-molecule distribution of a finite dataset and, therefore struggle to generate molecules with significant binding improvement over the training dataset. We instead frame the pocket-conditioned molecular generation task as an RL problem and develop TacoGFN, a target conditional Generative Flow Network model. Our method is explicitly encouraged to generate molecules with desired properties as opposed to fitting on a pre-existing data distribution. To this end, we develop transformer-based docking score prediction to speed up docking score computation and propose TacoGFN to explore molecule space efficiently. Furthermore, we incorporate several rounds of active learning where generated samples are queried using a docking oracle to improve the docking score prediction. This approach allows us to accurately explore as much of the molecule landscape as we can afford computationally. Empirically, molecules generated using TacoGFN and its variants significantly outperform all baseline methods across every property (Docking score, QED, SA, Lipinski), while being orders of magnitude faster.