 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePretraining Generative Flow Networks with Inexpensive Rewards for Molecular Graph Generation
Mar 08, 2025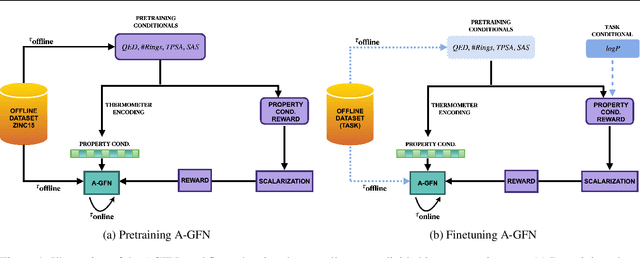



Generative Flow Networks (GFlowNets) have recently emerged as a suitable framework for generating diverse and high-quality molecular structures by learning from rewards treated as unnormalized distributions. Previous works in this framework often restrict exploration by using predefined molecular fragments as building blocks, limiting the chemical space that can be accessed. In this work, we introduce Atomic GFlowNets (A-GFNs), a foundational generative model leveraging individual atoms as building blocks to explore drug-like chemical space more comprehensively. We propose an unsupervised pre-training approach using drug-like molecule datasets, which teaches A-GFNs about inexpensive yet informative molecular descriptors such as drug-likeliness, topological polar surface area, and synthetic accessibility scores. These properties serve as proxy rewards, guiding A-GFNs towards regions of chemical space that exhibit desirable pharmacological properties. We further implement a goal-conditioned finetuning process, which adapts A-GFNs to optimize for specific target properties. In this work, we pretrain A-GFN on a subset of ZINC dataset, and by employing robust evaluation metrics we show the effectiveness of our approach when compared to other relevant baseline methods for a wide range of drug design tasks.
All SMILES Variational Autoencoder
Jun 03, 2019



Variational autoencoders (VAEs) defined over SMILES string and graph-based representations of molecules promise to improve the optimization of molecular properties, thereby revolutionizing the pharmaceuticals and materials industries. However, these VAEs are hindered by the non-unique nature of SMILES strings and the computational cost of graph convolutions. To efficiently pass messages along all paths through the molecular graph, we encode multiple SMILES strings of a single molecule using a set of stacked recurrent neural networks, pooling hidden representations of each atom between SMILES representations, and use attentional pooling to build a final fixed-length latent representation. By then decoding to a disjoint set of SMILES strings of the molecule, our All SMILES VAE learns an almost bijective mapping between molecules and latent representations near the high-probability-mass subspace of the prior. Our SMILES-derived but molecule-based latent representations significantly surpass the state-of-the-art in a variety of fully- and semi-supervised property regression and molecular property optimization tasks.
PADME: A Deep Learning-based Framework for Drug-Target Interaction Prediction
Oct 24, 2018

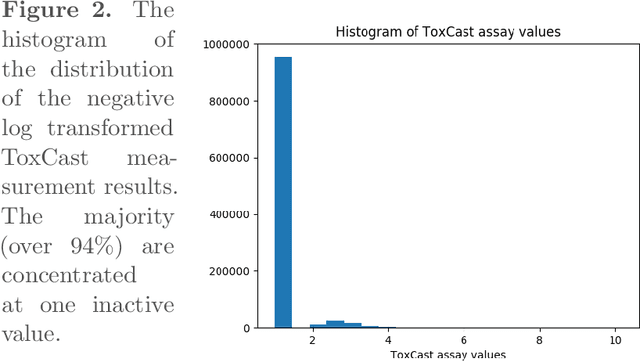

In silico drug-target interaction (DTI) prediction is an important and challenging problem in biomedical research with a huge potential benefit to the pharmaceutical industry and patients. Most existing methods for DTI prediction including deep learning models generally have binary endpoints, which could be an oversimplification of the problem, and those methods are typically unable to handle cold-target problems, i.e., problems involving target protein that never appeared in the training set. Towards this, we contrived PADME (Protein And Drug Molecule interaction prEdiction), a framework based on Deep Neural Networks, to predict real-valued interaction strength between compounds and proteins. PADME takes both compound and protein information as inputs, so it is capable of solving cold-target (and cold-drug) problems. To our knowledge, we are the first to combine Molecular Graph Convolution (MGC) for compound featurization with protein descriptors for DTI prediction. We used multiple cross-validation split schemes and evaluation metrics to measure the performance of PADME on multiple datasets, including the ToxCast dataset, which we believe should be a standard benchmark for DTI problems, and PADME consistently dominates baseline methods. The results of a case study, which predicts the interactions between various compounds and androgen receptor (AR), suggest PADME's potential in drug development. The scalability of PADME is another advantage in the age of Big Data.