 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeometric-informed GFlowNets for Structure-Based Drug Design
Paper and Code
Jun 16, 2024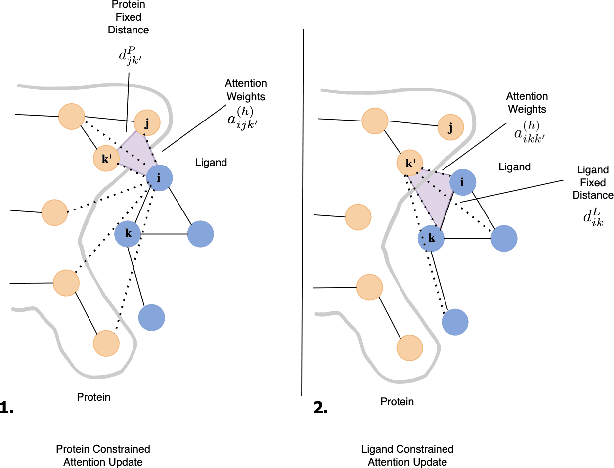


The rise of cost involved with drug discovery and current speed of which they are discover, underscore the need for more efficient structure-based drug design (SBDD) methods. We employ Generative Flow Networks (GFlowNets), to effectively explore the vast combinatorial space of drug-like molecules, which traditional virtual screening methods fail to cover. We introduce a novel modification to the GFlowNet framework by incorporating trigonometrically consistent embeddings, previously utilized in tasks involving protein conformation and protein-ligand interactions, to enhance the model's ability to generate molecules tailored to specific protein pockets. We have modified the existing protein conditioning used by GFlowNets, blending geometric information from both protein and ligand embeddings to achieve more geometrically consistent embeddings. Experiments conducted using CrossDocked2020 demonstrated an improvement in the binding affinity between generated molecules and protein pockets for both single and multi-objective tasks, compared to previous work. Additionally, we propose future work aimed at further increasing the geometric information captured in protein-ligand interactions.