 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTop-down machine learning of coarse-grained protein force-fields
Jun 27, 2023Developing accurate and efficient coarse-grained representations of proteins is crucial for understanding their folding, function, and interactions over extended timescales. Our methodology involves simulating proteins with molecular dynamics and utilizing the resulting trajectories to train a neural network potential through differentiable trajectory reweighting. Remarkably, this method requires only the native conformation of proteins, eliminating the need for labeled data derived from extensive simulations or memory-intensive end-to-end differentiable simulations. Once trained, the model can be employed to run parallel molecular dynamics simulations and sample folding events for proteins both within and beyond the training distribution, showcasing its extrapolation capabilities. By applying Markov State Models, native-like conformations of the simulated proteins can be predicted from the coarse-grained simulations. Owing to its theoretical transferability and ability to use solely experimental static structures as training data, we anticipate that this approach will prove advantageous for developing new protein force fields and further advancing the study of protein dynamics, folding, and interactions.
TensorNet: Cartesian Tensor Representations for Efficient Learning of Molecular Potentials
Jun 10, 2023The development of efficient machine learning models for molecular systems representation is becoming crucial in scientific research. We introduce TensorNet, an innovative $\mathrm{O}(3)$-equivariant message-passing neural network architecture that leverages Cartesian tensor representations. By using Cartesian tensor atomic embeddings, feature mixing is simplified through matrix product operations. Furthermore, the cost-effective decomposition of these tensors into rotation group irreducible representations allows for the separate processing of scalars, vectors, and tensors when necessary. Compared to higher-rank spherical tensor models, TensorNet demonstrates state-of-the-art performance with significantly fewer parameters. For small molecule potential energies, this can be achieved even with a single interaction layer. As a result of all these properties, the model's computational cost is substantially decreased. Moreover, the accurate prediction of vector and tensor molecular quantities on top of potential energies and forces is possible. In summary, TensorNet's framework opens up a new space for the design of state-of-the-art equivariant models.
Coarse Graining Molecular Dynamics with Graph Neural Networks
Aug 21, 2020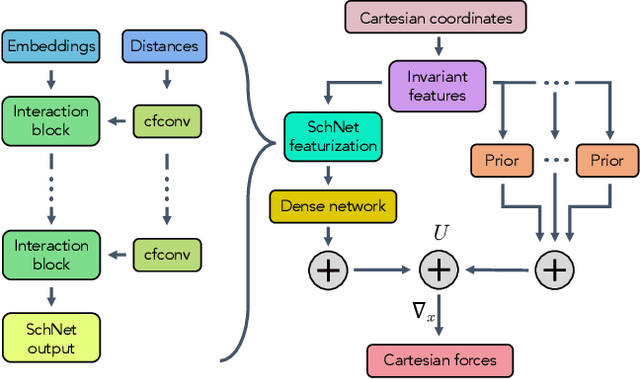
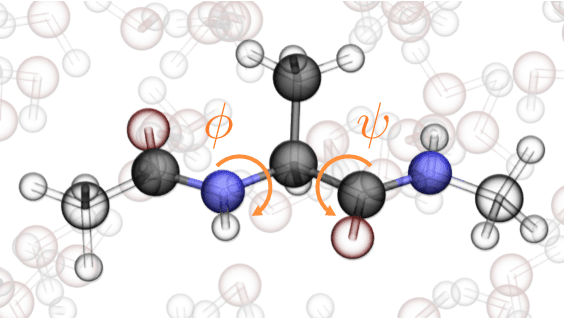
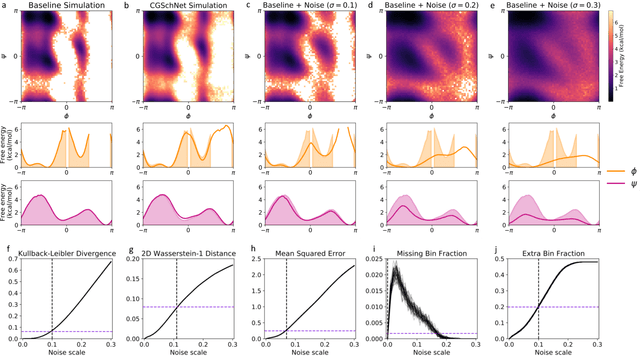
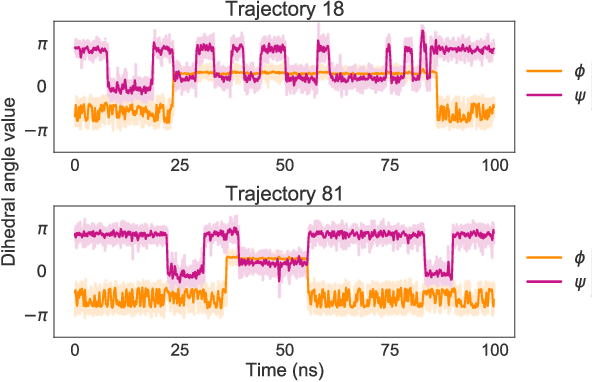
Coarse graining enables the investigation of molecular dynamics for larger systems and at longer timescales than is possible at atomic resolution. However, a coarse graining model must be formulated such that the conclusions we draw from it are consistent with the conclusions we would draw from a model at a finer level of detail. It has been proven that a force matching scheme defines a thermodynamically consistent coarse-grained model for an atomistic system in the variational limit. Wang et al. [ACS Cent. Sci. 5, 755 (2019)] demonstrated that the existence of such a variational limit enables the use of a supervised machine learning framework to generate a coarse-grained force field, which can then be used for simulation in the coarse-grained space. Their framework, however, requires the manual input of molecular features upon which to machine learn the force field. In the present contribution, we build upon the advance of Wang et al.and introduce a hybrid architecture for the machine learning of coarse-grained force fields that learns their own features via a subnetwork that leverages continuous filter convolutions on a graph neural network architecture. We demonstrate that this framework succeeds at reproducing the thermodynamics for small biomolecular systems. Since the learned molecular representations are inherently transferable, the architecture presented here sets the stage for the development of machine-learned, coarse-grained force fields that are transferable across molecular systems.