 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePlayMolecule pKAce: Small Molecule Protonation through Equivariant Neural Networks
Jul 15, 2024



Small molecule protonation is an important part of the preparation of small molecules for many types of computational chemistry protocols. For this, a correct estimation of the pKa values of the protonation sites of molecules is required. In this work, we present pKAce, a new web application for the prediction of micro-pKa values of the molecules' protonation sites. We adapt the state-of-the-art, equivariant, TensorNet model originally developed for quantum mechanics energy and force predictions to the prediction of micro-pKa values. We show that an adapted version of this model can achieve state-of-the-art performance comparable with established models while trained on just a fraction of their training data.
ACEGEN: Reinforcement learning of generative chemical agents for drug discovery
May 07, 2024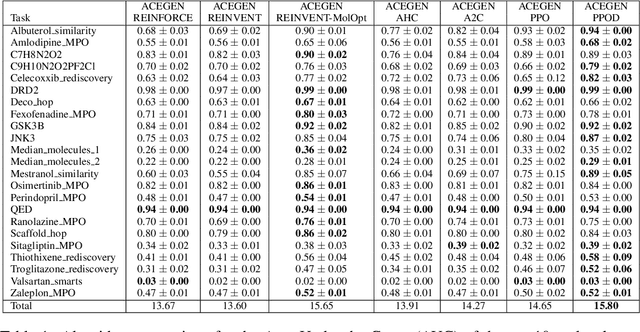


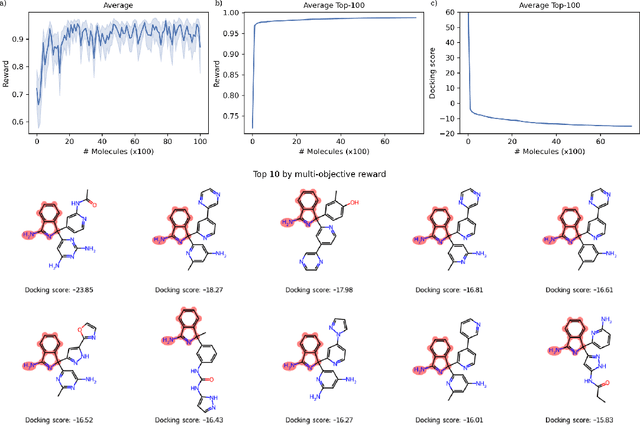
In recent years, reinforcement learning (RL) has emerged as a valuable tool in drug design, offering the potential to propose and optimize molecules with desired properties. However, striking a balance between capability, flexibility, and reliability remains challenging due to the complexity of advanced RL algorithms and the significant reliance on specialized code. In this work, we introduce ACEGEN, a comprehensive and streamlined toolkit tailored for generative drug design, built using TorchRL, a modern decision-making library that offers efficient and thoroughly tested reusable components. ACEGEN provides a robust, flexible, and efficient platform for molecular design. We validate its effectiveness by benchmarking it across various algorithms and conducting multiple drug discovery case studies. ACEGEN is accessible at https://github.com/acellera/acegen-open.
Navigating protein landscapes with a machine-learned transferable coarse-grained model
Oct 27, 2023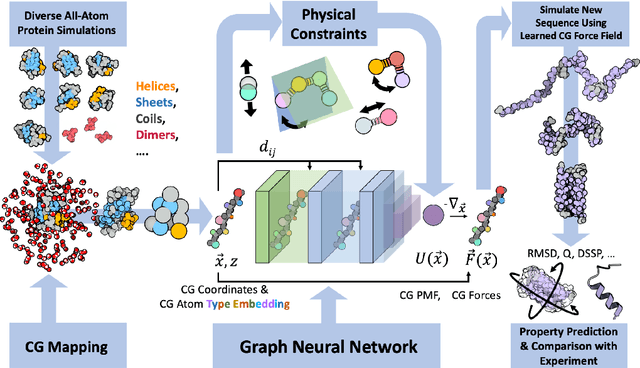
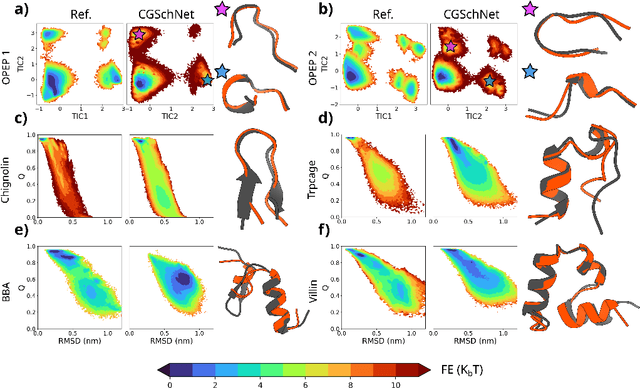

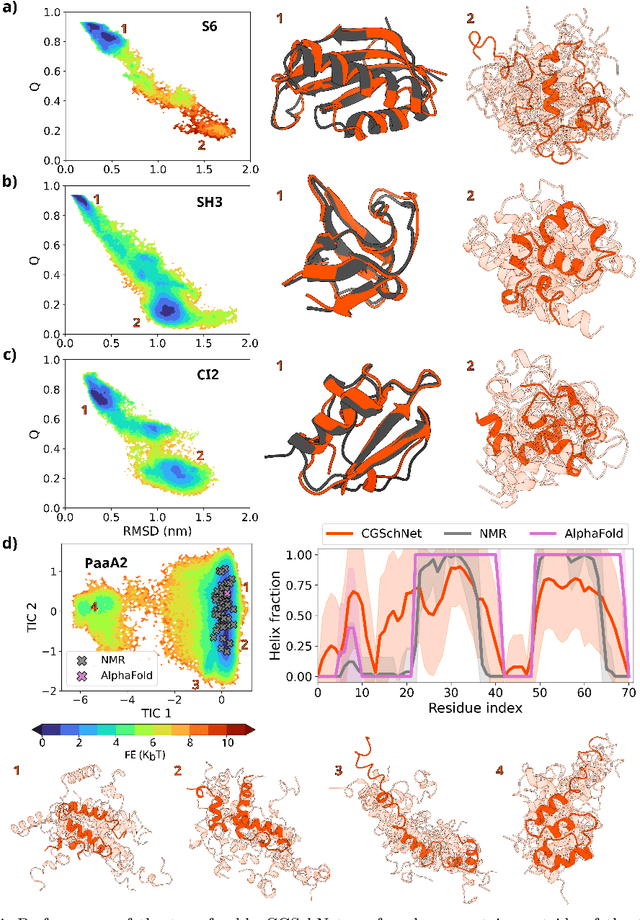
The most popular and universally predictive protein simulation models employ all-atom molecular dynamics (MD), but they come at extreme computational cost. The development of a universal, computationally efficient coarse-grained (CG) model with similar prediction performance has been a long-standing challenge. By combining recent deep learning methods with a large and diverse training set of all-atom protein simulations, we here develop a bottom-up CG force field with chemical transferability, which can be used for extrapolative molecular dynamics on new sequences not used during model parametrization. We demonstrate that the model successfully predicts folded structures, intermediates, metastable folded and unfolded basins, and the fluctuations of intrinsically disordered proteins while it is several orders of magnitude faster than an all-atom model. This showcases the feasibility of a universal and computationally efficient machine-learned CG model for proteins.
Machine Learning Small Molecule Properties in Drug Discovery
Aug 02, 2023



Machine learning (ML) is a promising approach for predicting small molecule properties in drug discovery. Here, we provide a comprehensive overview of various ML methods introduced for this purpose in recent years. We review a wide range of properties, including binding affinities, solubility, and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity). We discuss existing popular datasets and molecular descriptors and embeddings, such as chemical fingerprints and graph-based neural networks. We highlight also challenges of predicting and optimizing multiple properties during hit-to-lead and lead optimization stages of drug discovery and explore briefly possible multi-objective optimization techniques that can be used to balance diverse properties while optimizing lead candidates. Finally, techniques to provide an understanding of model predictions, especially for critical decision-making in drug discovery are assessed. Overall, this review provides insights into the landscape of ML models for small molecule property predictions in drug discovery. So far, there are multiple diverse approaches, but their performances are often comparable. Neural networks, while more flexible, do not always outperform simpler models. This shows that the availability of high-quality training data remains crucial for training accurate models and there is a need for standardized benchmarks, additional performance metrics, and best practices to enable richer comparisons between the different techniques and models that can shed a better light on the differences between the many techniques.
Top-down machine learning of coarse-grained protein force-fields
Jun 27, 2023Developing accurate and efficient coarse-grained representations of proteins is crucial for understanding their folding, function, and interactions over extended timescales. Our methodology involves simulating proteins with molecular dynamics and utilizing the resulting trajectories to train a neural network potential through differentiable trajectory reweighting. Remarkably, this method requires only the native conformation of proteins, eliminating the need for labeled data derived from extensive simulations or memory-intensive end-to-end differentiable simulations. Once trained, the model can be employed to run parallel molecular dynamics simulations and sample folding events for proteins both within and beyond the training distribution, showcasing its extrapolation capabilities. By applying Markov State Models, native-like conformations of the simulated proteins can be predicted from the coarse-grained simulations. Owing to its theoretical transferability and ability to use solely experimental static structures as training data, we anticipate that this approach will prove advantageous for developing new protein force fields and further advancing the study of protein dynamics, folding, and interactions.
Machine Learning Coarse-Grained Potentials of Protein Thermodynamics
Dec 14, 2022A generalized understanding of protein dynamics is an unsolved scientific problem, the solution of which is critical to the interpretation of the structure-function relationships that govern essential biological processes. Here, we approach this problem by constructing coarse-grained molecular potentials based on artificial neural networks and grounded in statistical mechanics. For training, we build a unique dataset of unbiased all-atom molecular dynamics simulations of approximately 9 ms for twelve different proteins with multiple secondary structure arrangements. The coarse-grained models are capable of accelerating the dynamics by more than three orders of magnitude while preserving the thermodynamics of the systems. Coarse-grained simulations identify relevant structural states in the ensemble with comparable energetics to the all-atom systems. Furthermore, we show that a single coarse-grained potential can integrate all twelve proteins and can capture experimental structural features of mutated proteins. These results indicate that machine learning coarse-grained potentials could provide a feasible approach to simulate and understand protein dynamics.