 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAceFF: A State-of-the-Art Machine Learning Potential for Small Molecules
Jan 02, 2026We introduce AceFF, a pre-trained machine learning interatomic potential (MLIP) optimized for small molecule drug discovery. While MLIPs have emerged as efficient alternatives to Density Functional Theory (DFT), generalizability across diverse chemical spaces remains difficult. AceFF addresses this via a refined TensorNet2 architecture trained on a comprehensive dataset of drug-like compounds. This approach yields a force field that balances high-throughput inference speed with DFT-level accuracy. AceFF fully supports the essential medicinal chemistry elements (H, B, C, N, O, F, Si, P, S, Cl, Br, I) and is explicitly trained to handle charged states. Validation against rigorous benchmarks, including complex torsional energy scans, molecular dynamics trajectories, batched minimizations, and forces and anergy accuracy demonstrates that AceFF establishes a new state-of-the-art for organic molecules. The AceFF-2 model weights and inference code are available at https://huggingface.co/Acellera/AceFF-2.0.
QuantumBind-RBFE: Accurate Relative Binding Free Energy Calculations Using Neural Network Potentials
Jan 03, 2025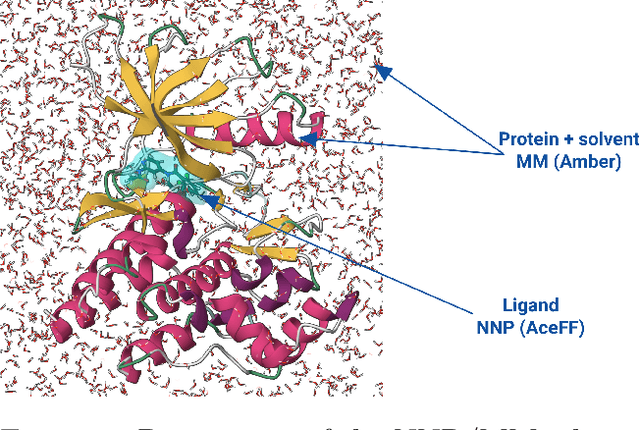
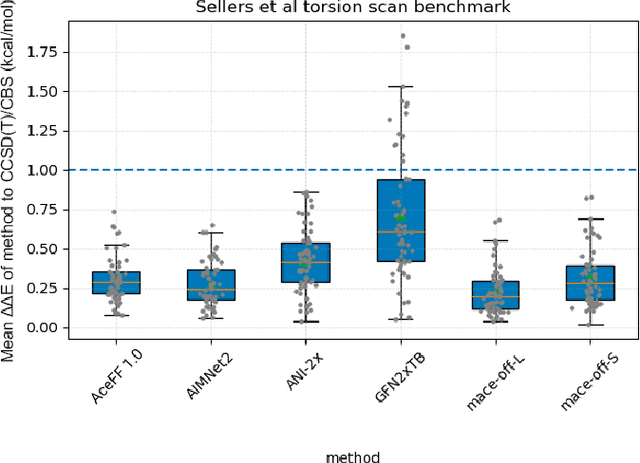


Accurate prediction of protein-ligand binding affinities is crucial in drug discovery, particularly during hit-to-lead and lead optimization phases, however, limitations in ligand force fields continue to impact prediction accuracy. In this work, we validate relative binding free energy (RBFE) accuracy using neural network potentials (NNPs) for the ligands. We utilize a novel NNP model, AceForce 1.0, based on the TensorNet architecture for small molecules that broadens the applicability to diverse drug-like compounds, including all important chemical elements and supporting charged molecules. Using established benchmarks, we show overall improved accuracy and correlation in binding affinity predictions compared with GAFF2 for molecular mechanics and ANI2-x for NNPs. Slightly less accuracy but comparable correlations with OPLS4. We also show that we can run the NNP simulations at 2 fs timestep, at least two times larger than previous NNP models, providing significant speed gains. The results show promise for further evolutions of free energy calculations using NNPs while demonstrating its practical use already with the current generation. The code and NNP model are publicly available for research use.
TorchMD-Net 2.0: Fast Neural Network Potentials for Molecular Simulations
Feb 27, 2024



Achieving a balance between computational speed, prediction accuracy, and universal applicability in molecular simulations has been a persistent challenge. This paper presents substantial advancements in the TorchMD-Net software, a pivotal step forward in the shift from conventional force fields to neural network-based potentials. The evolution of TorchMD-Net into a more comprehensive and versatile framework is highlighted, incorporating cutting-edge architectures such as TensorNet. This transformation is achieved through a modular design approach, encouraging customized applications within the scientific community. The most notable enhancement is a significant improvement in computational efficiency, achieving a very remarkable acceleration in the computation of energy and forces for TensorNet models, with performance gains ranging from 2-fold to 10-fold over previous iterations. Other enhancements include highly optimized neighbor search algorithms that support periodic boundary conditions and the smooth integration with existing molecular dynamics frameworks. Additionally, the updated version introduces the capability to integrate physical priors, further enriching its application spectrum and utility in research. The software is available at https://github.com/torchmd/torchmd-net.
Machine Learning Coarse-Grained Potentials of Protein Thermodynamics
Dec 14, 2022A generalized understanding of protein dynamics is an unsolved scientific problem, the solution of which is critical to the interpretation of the structure-function relationships that govern essential biological processes. Here, we approach this problem by constructing coarse-grained molecular potentials based on artificial neural networks and grounded in statistical mechanics. For training, we build a unique dataset of unbiased all-atom molecular dynamics simulations of approximately 9 ms for twelve different proteins with multiple secondary structure arrangements. The coarse-grained models are capable of accelerating the dynamics by more than three orders of magnitude while preserving the thermodynamics of the systems. Coarse-grained simulations identify relevant structural states in the ensemble with comparable energetics to the all-atom systems. Furthermore, we show that a single coarse-grained potential can integrate all twelve proteins and can capture experimental structural features of mutated proteins. These results indicate that machine learning coarse-grained potentials could provide a feasible approach to simulate and understand protein dynamics.
NNP/MM: Fast molecular dynamics simulations with machine learning potentials and molecular mechanics
Jan 20, 2022
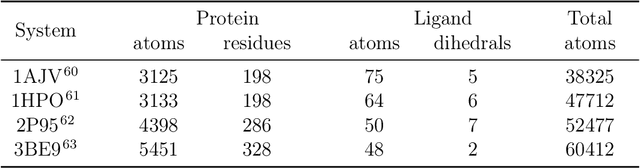
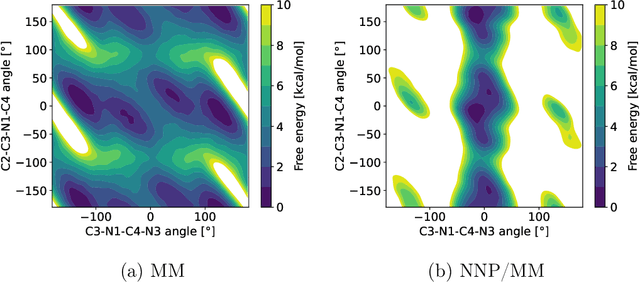
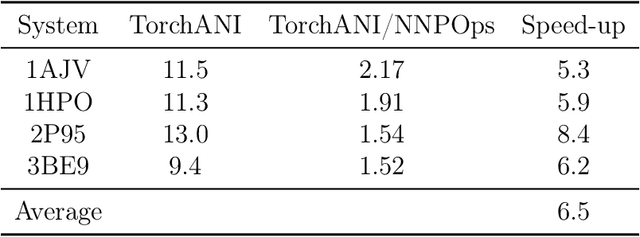
Parametric and non-parametric machine learning potentials have emerged recently as a way to improve the accuracy of bio-molecular simulations. Here, we present NNP/MM, an hybrid method integrating neural network potentials (NNPs) and molecular mechanics (MM). It allows to simulate a part of molecular system with NNP, while the rest is simulated with MM for efficiency. The method is currently available in ACEMD using OpenMM plugins to optimize the performance of NNPs. The achieved performance is slower but comparable to the state-of-the-art GPU-accelerated MM simulations. We validated NNP/MM by performing MD simulations of four protein-ligand complexes, where NNP is used for the intra-molecular interactions of a lignad and MM for the rest interactions. This shows that NNP can already replace MM for small molecules in protein-ligand simulations. The combined sampling of each complex is 1 microsecond, which are the longest simulations of NNP/MM ever reported. Finally, we have made the setup of the NNP/MM simulations simple and user-friendly.
TorchMD: A deep learning framework for molecular simulations
Dec 22, 2020


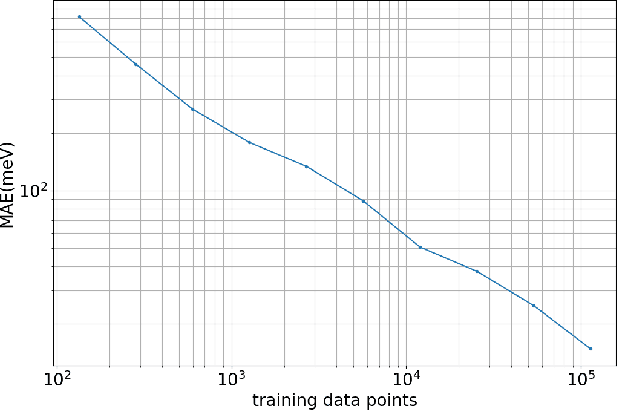
Molecular dynamics simulations provide a mechanistic description of molecules by relying on empirical potentials. The quality and transferability of such potentials can be improved leveraging data-driven models derived with machine learning approaches. Here, we present TorchMD, a framework for molecular simulations with mixed classical and machine learning potentials. All of force computations including bond, angle, dihedral, Lennard-Jones and Coulomb interactions are expressed as PyTorch arrays and operations. Moreover, TorchMD enables learning and simulating neural network potentials. We validate it using standard Amber all-atom simulations, learning an ab-initio potential, performing an end-to-end training and finally learning and simulating a coarse-grained model for protein folding. We believe that TorchMD provides a useful tool-set to support molecular simulations of machine learning potentials. Code and data are freely available at \url{github.com/torchmd}.
Dimensionality reduction methods for molecular simulations
Nov 02, 2017
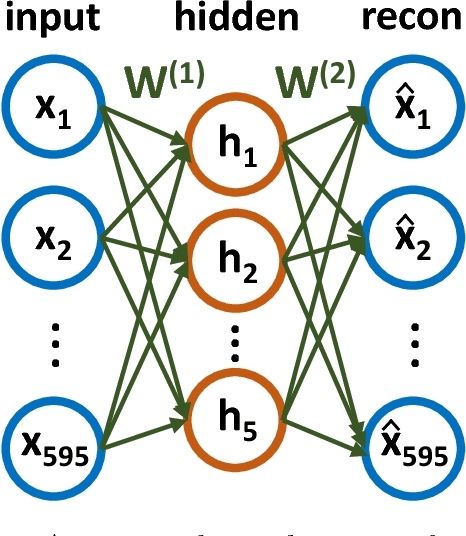
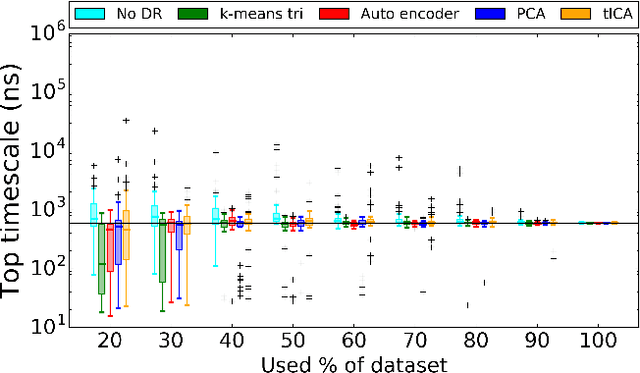
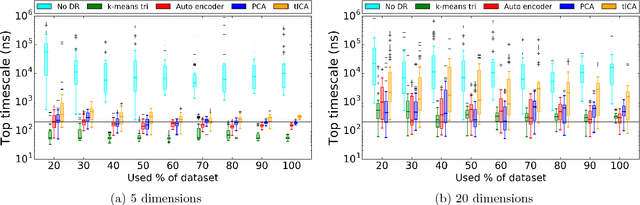
Molecular simulations produce very high-dimensional data-sets with millions of data points. As analysis methods are often unable to cope with so many dimensions, it is common to use dimensionality reduction and clustering methods to reach a reduced representation of the data. Yet these methods often fail to capture the most important features necessary for the construction of a Markov model. Here we demonstrate the results of various dimensionality reduction methods on two simulation data-sets, one of protein folding and another of protein-ligand binding. The methods tested include a k-means clustering variant, a non-linear auto encoder, principal component analysis and tICA. The dimension-reduced data is then used to estimate the implied timescales of the slowest process by a Markov state model analysis to assess the quality of the projection. The projected dimensions learned from the data are visualized to demonstrate which conformations the various methods choose to represent the molecular process.