 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAceFF: A State-of-the-Art Machine Learning Potential for Small Molecules
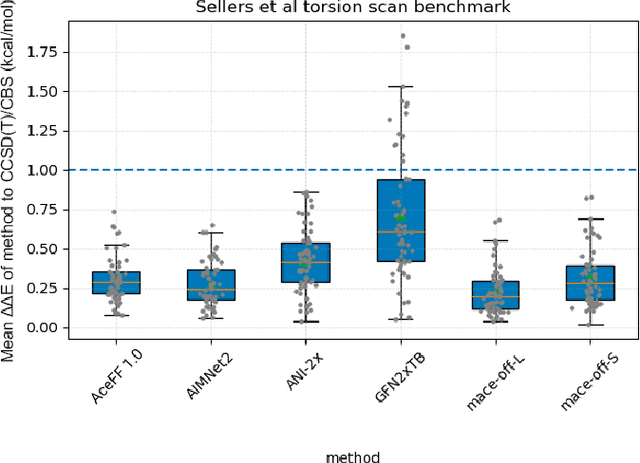
Jan 02, 2026We introduce AceFF, a pre-trained machine learning interatomic potential (MLIP) optimized for small molecule drug discovery. While MLIPs have emerged as efficient alternatives to Density Functional Theory (DFT), generalizability across diverse chemical spaces remains difficult. AceFF addresses this via a refined TensorNet2 architecture trained on a comprehensive dataset of drug-like compounds. This approach yields a force field that balances high-throughput inference speed with DFT-level accuracy. AceFF fully supports the essential medicinal chemistry elements (H, B, C, N, O, F, Si, P, S, Cl, Br, I) and is explicitly trained to handle charged states. Validation against rigorous benchmarks, including complex torsional energy scans, molecular dynamics trajectories, batched minimizations, and forces and anergy accuracy demonstrates that AceFF establishes a new state-of-the-art for organic molecules. The AceFF-2 model weights and inference code are available at https://huggingface.co/Acellera/AceFF-2.0.
Test-Time Training Scaling for Chemical Exploration in Drug Design
Jan 31, 2025



Chemical language models for molecular design have the potential to find solutions to multi-parameter optimization problems in drug discovery via reinforcement learning (RL). A key requirement to achieve this is the capacity to "search" chemical space to identify all molecules of interest. Here, we propose a challenging new benchmark to discover dissimilar molecules that possess similar bioactivity, a common scenario in drug discovery, but a hard problem to optimize. We show that a population of RL agents can solve the benchmark, while a single agent cannot. We also find that cooperative strategies are not significantly better than independent agents. Moreover, the performance on the benchmark scales log-linearly with the number of independent agents, showing a test-time training scaling law for chemical language models.
REINFORCE-ING Chemical Language Models in Drug Design
Jan 27, 2025



Chemical language models, combined with reinforcement learning, have shown significant promise to efficiently traverse large chemical spaces in drug design. However, the performance of various RL algorithms and their best practices for practical drug design are still unclear. Here, starting from the principles of the REINFORCE algorithm, we investigate the effect of different components from RL theory including experience replay, hill-climbing, baselines to reduce variance, and alternative reward shaping. Additionally we demonstrate how RL hyperparameters can be fine-tuned for effectiveness, efficiency, or chemical regularization as demonstrated using the MolOpt benchmark.
QuantumBind-RBFE: Accurate Relative Binding Free Energy Calculations Using Neural Network Potentials
Jan 03, 2025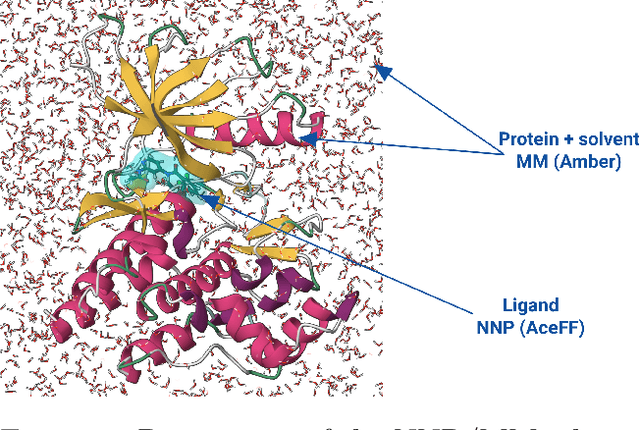


Accurate prediction of protein-ligand binding affinities is crucial in drug discovery, particularly during hit-to-lead and lead optimization phases, however, limitations in ligand force fields continue to impact prediction accuracy. In this work, we validate relative binding free energy (RBFE) accuracy using neural network potentials (NNPs) for the ligands. We utilize a novel NNP model, AceForce 1.0, based on the TensorNet architecture for small molecules that broadens the applicability to diverse drug-like compounds, including all important chemical elements and supporting charged molecules. Using established benchmarks, we show overall improved accuracy and correlation in binding affinity predictions compared with GAFF2 for molecular mechanics and ANI2-x for NNPs. Slightly less accuracy but comparable correlations with OPLS4. We also show that we can run the NNP simulations at 2 fs timestep, at least two times larger than previous NNP models, providing significant speed gains. The results show promise for further evolutions of free energy calculations using NNPs while demonstrating its practical use already with the current generation. The code and NNP model are publicly available for research use.
AMARO: All Heavy-Atom Transferable Neural Network Potentials of Protein Thermodynamics
Sep 26, 2024



All-atom molecular simulations offer detailed insights into macromolecular phenomena, but their substantial computational cost hinders the exploration of complex biological processes. We introduce Advanced Machine-learning Atomic Representation Omni-force-field (AMARO), a new neural network potential (NNP) that combines an O(3)-equivariant message-passing neural network architecture, TensorNet, with a coarse-graining map that excludes hydrogen atoms. AMARO demonstrates the feasibility of training coarser NNP, without prior energy terms, to run stable protein dynamics with scalability and generalization capabilities.
PlayMolecule pKAce: Small Molecule Protonation through Equivariant Neural Networks
Jul 15, 2024



Small molecule protonation is an important part of the preparation of small molecules for many types of computational chemistry protocols. For this, a correct estimation of the pKa values of the protonation sites of molecules is required. In this work, we present pKAce, a new web application for the prediction of micro-pKa values of the molecules' protonation sites. We adapt the state-of-the-art, equivariant, TensorNet model originally developed for quantum mechanics energy and force predictions to the prediction of micro-pKa values. We show that an adapted version of this model can achieve state-of-the-art performance comparable with established models while trained on just a fraction of their training data.
BricksRL: A Platform for Democratizing Robotics and Reinforcement Learning Research and Education with LEGO
Jun 25, 2024



We present BricksRL, a platform designed to democratize access to robotics for reinforcement learning research and education. BricksRL facilitates the creation, design, and training of custom LEGO robots in the real world by interfacing them with the TorchRL library for reinforcement learning agents. The integration of TorchRL with the LEGO hubs, via Bluetooth bidirectional communication, enables state-of-the-art reinforcement learning training on GPUs for a wide variety of LEGO builds. This offers a flexible and cost-efficient approach for scaling and also provides a robust infrastructure for robot-environment-algorithm communication. We present various experiments across tasks and robot configurations, providing built plans and training results. Furthermore, we demonstrate that inexpensive LEGO robots can be trained end-to-end in the real world to achieve simple tasks, with training times typically under 120 minutes on a normal laptop. Moreover, we show how users can extend the capabilities, exemplified by the successful integration of non-LEGO sensors. By enhancing accessibility to both robotics and reinforcement learning, BricksRL establishes a strong foundation for democratized robotic learning in research and educational settings.
ACEGEN: Reinforcement learning of generative chemical agents for drug discovery
May 07, 2024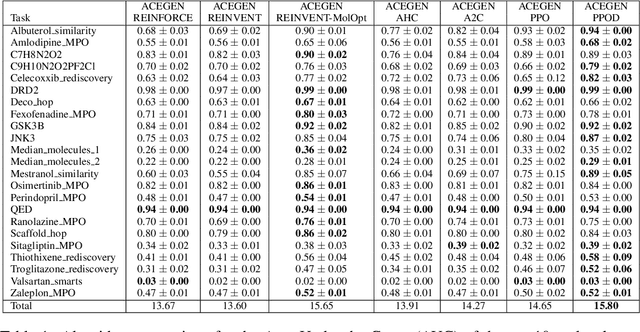


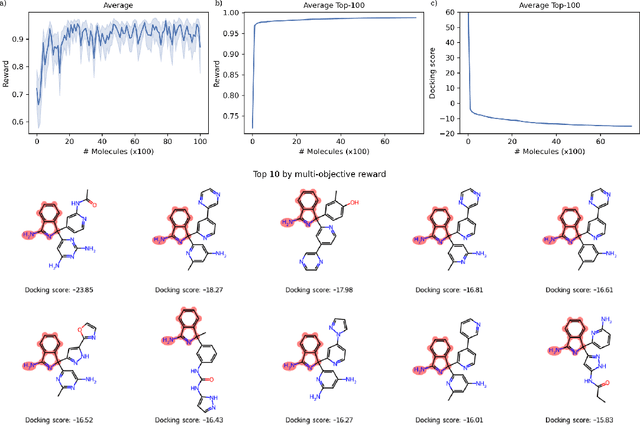
In recent years, reinforcement learning (RL) has emerged as a valuable tool in drug design, offering the potential to propose and optimize molecules with desired properties. However, striking a balance between capability, flexibility, and reliability remains challenging due to the complexity of advanced RL algorithms and the significant reliance on specialized code. In this work, we introduce ACEGEN, a comprehensive and streamlined toolkit tailored for generative drug design, built using TorchRL, a modern decision-making library that offers efficient and thoroughly tested reusable components. ACEGEN provides a robust, flexible, and efficient platform for molecular design. We validate its effectiveness by benchmarking it across various algorithms and conducting multiple drug discovery case studies. ACEGEN is accessible at https://github.com/acellera/acegen-open.
On the Inclusion of Charge and Spin States in Cartesian Tensor Neural Network Potentials
Mar 22, 2024



In this letter, we present an extension to TensorNet, a state-of-the-art equivariant Cartesian tensor neural network potential, allowing it to handle charged molecules and spin states without architectural changes or increased costs. By incorporating these attributes, we address input degeneracy issues, enhancing the model's predictive accuracy across diverse chemical systems. This advancement significantly broadens TensorNet's applicability, maintaining its efficiency and accuracy.
TorchMD-Net 2.0: Fast Neural Network Potentials for Molecular Simulations
Feb 27, 2024



Achieving a balance between computational speed, prediction accuracy, and universal applicability in molecular simulations has been a persistent challenge. This paper presents substantial advancements in the TorchMD-Net software, a pivotal step forward in the shift from conventional force fields to neural network-based potentials. The evolution of TorchMD-Net into a more comprehensive and versatile framework is highlighted, incorporating cutting-edge architectures such as TensorNet. This transformation is achieved through a modular design approach, encouraging customized applications within the scientific community. The most notable enhancement is a significant improvement in computational efficiency, achieving a very remarkable acceleration in the computation of energy and forces for TensorNet models, with performance gains ranging from 2-fold to 10-fold over previous iterations. Other enhancements include highly optimized neighbor search algorithms that support periodic boundary conditions and the smooth integration with existing molecular dynamics frameworks. Additionally, the updated version introduces the capability to integrate physical priors, further enriching its application spectrum and utility in research. The software is available at https://github.com/torchmd/torchmd-net.