 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAceFF: A State-of-the-Art Machine Learning Potential for Small Molecules
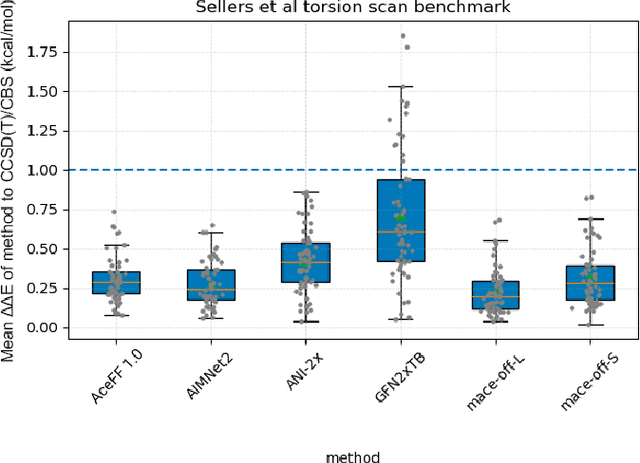
Jan 02, 2026We introduce AceFF, a pre-trained machine learning interatomic potential (MLIP) optimized for small molecule drug discovery. While MLIPs have emerged as efficient alternatives to Density Functional Theory (DFT), generalizability across diverse chemical spaces remains difficult. AceFF addresses this via a refined TensorNet2 architecture trained on a comprehensive dataset of drug-like compounds. This approach yields a force field that balances high-throughput inference speed with DFT-level accuracy. AceFF fully supports the essential medicinal chemistry elements (H, B, C, N, O, F, Si, P, S, Cl, Br, I) and is explicitly trained to handle charged states. Validation against rigorous benchmarks, including complex torsional energy scans, molecular dynamics trajectories, batched minimizations, and forces and anergy accuracy demonstrates that AceFF establishes a new state-of-the-art for organic molecules. The AceFF-2 model weights and inference code are available at https://huggingface.co/Acellera/AceFF-2.0.
QuantumBind-RBFE: Accurate Relative Binding Free Energy Calculations Using Neural Network Potentials
Jan 03, 2025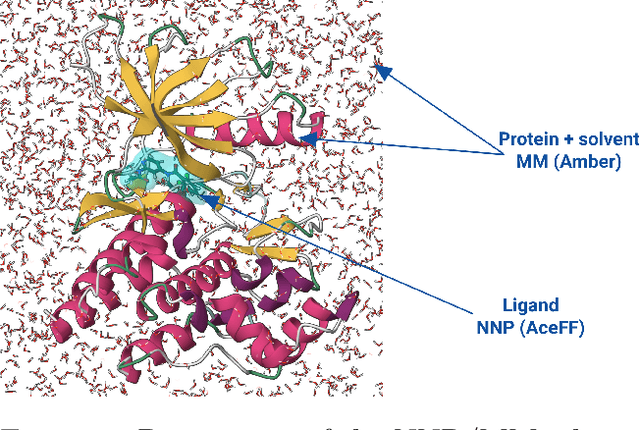


Accurate prediction of protein-ligand binding affinities is crucial in drug discovery, particularly during hit-to-lead and lead optimization phases, however, limitations in ligand force fields continue to impact prediction accuracy. In this work, we validate relative binding free energy (RBFE) accuracy using neural network potentials (NNPs) for the ligands. We utilize a novel NNP model, AceForce 1.0, based on the TensorNet architecture for small molecules that broadens the applicability to diverse drug-like compounds, including all important chemical elements and supporting charged molecules. Using established benchmarks, we show overall improved accuracy and correlation in binding affinity predictions compared with GAFF2 for molecular mechanics and ANI2-x for NNPs. Slightly less accuracy but comparable correlations with OPLS4. We also show that we can run the NNP simulations at 2 fs timestep, at least two times larger than previous NNP models, providing significant speed gains. The results show promise for further evolutions of free energy calculations using NNPs while demonstrating its practical use already with the current generation. The code and NNP model are publicly available for research use.
OpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials
Oct 04, 2023



Machine learning plays an important and growing role in molecular simulation. The newest version of the OpenMM molecular dynamics toolkit introduces new features to support the use of machine learning potentials. Arbitrary PyTorch models can be added to a simulation and used to compute forces and energy. A higher-level interface allows users to easily model their molecules of interest with general purpose, pretrained potential functions. A collection of optimized CUDA kernels and custom PyTorch operations greatly improves the speed of simulations. We demonstrate these features on simulations of cyclin-dependent kinase 8 (CDK8) and the green fluorescent protein (GFP) chromophore in water. Taken together, these features make it practical to use machine learning to improve the accuracy of simulations at only a modest increase in cost.