 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePushing the limits of one-dimensional NMR spectroscopy for automated structure elucidation using artificial intelligence
Dec 20, 2025One-dimensional NMR spectroscopy is one of the most widely used techniques for the characterization of organic compounds and natural products. For molecules with up to 36 non-hydrogen atoms, the number of possible structures has been estimated to range from $10^{20} - 10^{60}$. The task of determining the structure (formula and connectivity) of a molecule of this size using only its one-dimensional $^1$H and/or $^{13}$C NMR spectrum, i.e. de novo structure generation, thus appears completely intractable. Here we show how it is possible to achieve this task for systems with up to 40 non-hydrogen atoms across the full elemental coverage typically encountered in organic chemistry (C, N, O, H, P, S, Si, B, and the halogens) using a deep learning framework, thus covering a vast portion of the drug-like chemical space. Leveraging insights from natural language processing, we show that our transformer-based architecture predicts the correct molecule with 55.2% accuracy within the first 15 predictions using only the $^1$H and $^{13}$C NMR spectra, thus overcoming the combinatorial growth of the chemical space while also being extensible to experimental data via fine-tuning.
Accurate and efficient structure elucidation from routine one-dimensional NMR spectra using multitask machine learning
Aug 15, 2024Rapid determination of molecular structures can greatly accelerate workflows across many chemical disciplines. However, elucidating structure using only one-dimensional (1D) NMR spectra, the most readily accessible data, remains an extremely challenging problem because of the combinatorial explosion of the number of possible molecules as the number of constituent atoms is increased. Here, we introduce a multitask machine learning framework that predicts the molecular structure (formula and connectivity) of an unknown compound solely based on its 1D 1H and/or 13C NMR spectra. First, we show how a transformer architecture can be constructed to efficiently solve the task, traditionally performed by chemists, of assembling large numbers of molecular fragments into molecular structures. Integrating this capability with a convolutional neural network (CNN), we build an end-to-end model for predicting structure from spectra that is fast and accurate. We demonstrate the effectiveness of this framework on molecules with up to 19 heavy (non-hydrogen) atoms, a size for which there are trillions of possible structures. Without relying on any prior chemical knowledge such as the molecular formula, we show that our approach predicts the exact molecule 69.6% of the time within the first 15 predictions, reducing the search space by up to 11 orders of magnitude.
Nutmeg and SPICE: Models and Data for Biomolecular Machine Learning
Jun 18, 2024



We describe version 2 of the SPICE dataset, a collection of quantum chemistry calculations for training machine learning potentials. It expands on the original dataset by adding much more sampling of chemical space and more data on non-covalent interactions. We train a set of potential energy functions called Nutmeg on it. They use a novel mechanism to improve performance on charged and polar molecules, injecting precomputed partial charges into the model to provide a reference for the large scale charge distribution. Evaluation of the new models shows they do an excellent job of reproducing energy differences between conformations, even on highly charged molecules or ones that are significantly larger than the molecules in the training set. They also produce stable molecular dynamics trajectories, and are fast enough to be useful for routine simulation of small molecules.
TorchMD-Net 2.0: Fast Neural Network Potentials for Molecular Simulations
Feb 27, 2024



Achieving a balance between computational speed, prediction accuracy, and universal applicability in molecular simulations has been a persistent challenge. This paper presents substantial advancements in the TorchMD-Net software, a pivotal step forward in the shift from conventional force fields to neural network-based potentials. The evolution of TorchMD-Net into a more comprehensive and versatile framework is highlighted, incorporating cutting-edge architectures such as TensorNet. This transformation is achieved through a modular design approach, encouraging customized applications within the scientific community. The most notable enhancement is a significant improvement in computational efficiency, achieving a very remarkable acceleration in the computation of energy and forces for TensorNet models, with performance gains ranging from 2-fold to 10-fold over previous iterations. Other enhancements include highly optimized neighbor search algorithms that support periodic boundary conditions and the smooth integration with existing molecular dynamics frameworks. Additionally, the updated version introduces the capability to integrate physical priors, further enriching its application spectrum and utility in research. The software is available at https://github.com/torchmd/torchmd-net.
OpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials
Oct 04, 2023



Machine learning plays an important and growing role in molecular simulation. The newest version of the OpenMM molecular dynamics toolkit introduces new features to support the use of machine learning potentials. Arbitrary PyTorch models can be added to a simulation and used to compute forces and energy. A higher-level interface allows users to easily model their molecules of interest with general purpose, pretrained potential functions. A collection of optimized CUDA kernels and custom PyTorch operations greatly improves the speed of simulations. We demonstrate these features on simulations of cyclin-dependent kinase 8 (CDK8) and the green fluorescent protein (GFP) chromophore in water. Taken together, these features make it practical to use machine learning to improve the accuracy of simulations at only a modest increase in cost.
SPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials
Sep 21, 2022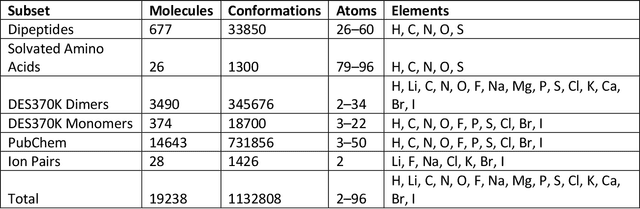
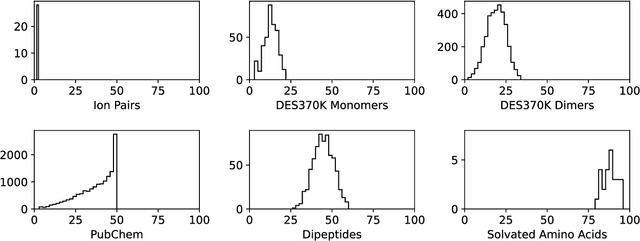

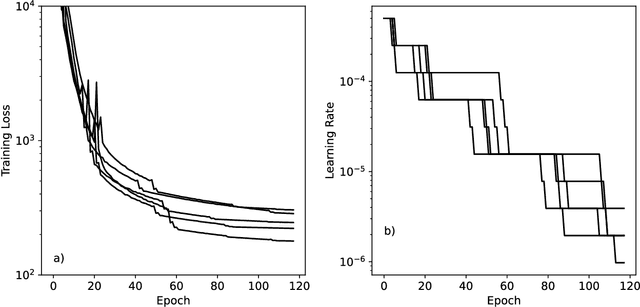
Machine learning potentials are an important tool for molecular simulation, but their development is held back by a shortage of high quality datasets to train them on. We describe the SPICE dataset, a new quantum chemistry dataset for training potentials relevant to simulating drug-like small molecules interacting with proteins. It contains over 1.1 million conformations for a diverse set of small molecules, dimers, dipeptides, and solvated amino acids. It includes 15 elements, charged and uncharged molecules, and a wide range of covalent and non-covalent interactions. It provides both forces and energies calculated at the {\omega}B97M-D3(BJ)/def2-TZVPPD level of theory, along with other useful quantities such as multipole moments and bond orders. We train a set of machine learning potentials on it and demonstrate that they can achieve chemical accuracy across a broad region of chemical space. It can serve as a valuable resource for the creation of transferable, ready to use potential functions for use in molecular simulations.
NNP/MM: Fast molecular dynamics simulations with machine learning potentials and molecular mechanics
Jan 20, 2022Parametric and non-parametric machine learning potentials have emerged recently as a way to improve the accuracy of bio-molecular simulations. Here, we present NNP/MM, an hybrid method integrating neural network potentials (NNPs) and molecular mechanics (MM). It allows to simulate a part of molecular system with NNP, while the rest is simulated with MM for efficiency. The method is currently available in ACEMD using OpenMM plugins to optimize the performance of NNPs. The achieved performance is slower but comparable to the state-of-the-art GPU-accelerated MM simulations. We validated NNP/MM by performing MD simulations of four protein-ligand complexes, where NNP is used for the intra-molecular interactions of a lignad and MM for the rest interactions. This shows that NNP can already replace MM for small molecules in protein-ligand simulations. The combined sampling of each complex is 1 microsecond, which are the longest simulations of NNP/MM ever reported. Finally, we have made the setup of the NNP/MM simulations simple and user-friendly.