 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNutmeg and SPICE: Models and Data for Biomolecular Machine Learning
Jun 18, 2024



We describe version 2 of the SPICE dataset, a collection of quantum chemistry calculations for training machine learning potentials. It expands on the original dataset by adding much more sampling of chemical space and more data on non-covalent interactions. We train a set of potential energy functions called Nutmeg on it. They use a novel mechanism to improve performance on charged and polar molecules, injecting precomputed partial charges into the model to provide a reference for the large scale charge distribution. Evaluation of the new models shows they do an excellent job of reproducing energy differences between conformations, even on highly charged molecules or ones that are significantly larger than the molecules in the training set. They also produce stable molecular dynamics trajectories, and are fast enough to be useful for routine simulation of small molecules.
SPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials
Sep 21, 2022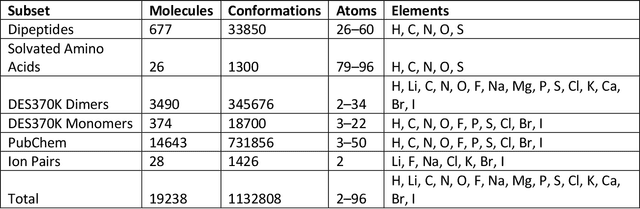
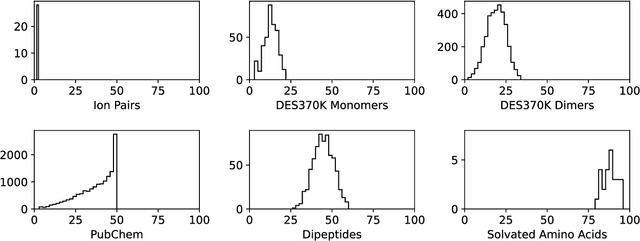

Machine learning potentials are an important tool for molecular simulation, but their development is held back by a shortage of high quality datasets to train them on. We describe the SPICE dataset, a new quantum chemistry dataset for training potentials relevant to simulating drug-like small molecules interacting with proteins. It contains over 1.1 million conformations for a diverse set of small molecules, dimers, dipeptides, and solvated amino acids. It includes 15 elements, charged and uncharged molecules, and a wide range of covalent and non-covalent interactions. It provides both forces and energies calculated at the {\omega}B97M-D3(BJ)/def2-TZVPPD level of theory, along with other useful quantities such as multipole moments and bond orders. We train a set of machine learning potentials on it and demonstrate that they can achieve chemical accuracy across a broad region of chemical space. It can serve as a valuable resource for the creation of transferable, ready to use potential functions for use in molecular simulations.