 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNutmeg and SPICE: Models and Data for Biomolecular Machine Learning
Jun 18, 2024



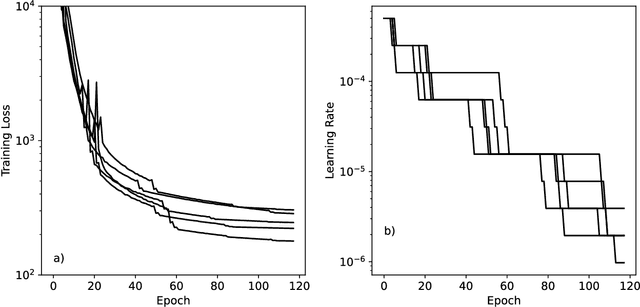
We describe version 2 of the SPICE dataset, a collection of quantum chemistry calculations for training machine learning potentials. It expands on the original dataset by adding much more sampling of chemical space and more data on non-covalent interactions. We train a set of potential energy functions called Nutmeg on it. They use a novel mechanism to improve performance on charged and polar molecules, injecting precomputed partial charges into the model to provide a reference for the large scale charge distribution. Evaluation of the new models shows they do an excellent job of reproducing energy differences between conformations, even on highly charged molecules or ones that are significantly larger than the molecules in the training set. They also produce stable molecular dynamics trajectories, and are fast enough to be useful for routine simulation of small molecules.
OpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials
Oct 04, 2023



Machine learning plays an important and growing role in molecular simulation. The newest version of the OpenMM molecular dynamics toolkit introduces new features to support the use of machine learning potentials. Arbitrary PyTorch models can be added to a simulation and used to compute forces and energy. A higher-level interface allows users to easily model their molecules of interest with general purpose, pretrained potential functions. A collection of optimized CUDA kernels and custom PyTorch operations greatly improves the speed of simulations. We demonstrate these features on simulations of cyclin-dependent kinase 8 (CDK8) and the green fluorescent protein (GFP) chromophore in water. Taken together, these features make it practical to use machine learning to improve the accuracy of simulations at only a modest increase in cost.
Espaloma-0.3.0: Machine-learned molecular mechanics force field for the simulation of protein-ligand systems and beyond
Jul 13, 2023



Molecular mechanics (MM) force fields -- the models that characterize the energy landscape of molecular systems via simple pairwise and polynomial terms -- have traditionally relied on human expert-curated, inflexible, and poorly extensible discrete chemical parameter assignment rules, namely atom or valence types. Recently, there has been significant interest in using graph neural networks to replace this process, while enabling the parametrization scheme to be learned in an end-to-end differentiable manner directly from quantum chemical calculations or condensed-phase data. In this paper, we extend the Espaloma end-to-end differentiable force field construction approach by incorporating both energy and force fitting directly to quantum chemical data into the training process. Building on the OpenMM SPICE dataset, we curate a dataset containing chemical spaces highly relevant to the broad interest of biomolecular modeling, covering small molecules, proteins, and RNA. The resulting force field, espaloma 0.3.0, self-consistently parametrizes these diverse biomolecular species, accurately predicts quantum chemical energies and forces, and maintains stable quantum chemical energy-minimized geometries. Surprisingly, this simple approach produces highly accurate protein-ligand binding free energies when self-consistently parametrizing protein and ligand. This approach -- capable of fitting new force fields to large quantum chemical datasets in one GPU-day -- shows significant promise as a path forward for building systematically more accurate force fields that can be easily extended to new chemical domains of interest.
EspalomaCharge: Machine learning-enabled ultra-fast partial charge assignment
Feb 16, 2023Atomic partial charges are crucial parameters in molecular dynamics (MD) simulation, dictating the electrostatic contributions to intermolecular energies, and thereby the potential energy landscape. Traditionally, the assignment of partial charges has relied on surrogates of \textit{ab initio} semiempirical quantum chemical methods such as AM1-BCC, and is expensive for large systems or large numbers of molecules. We propose a hybrid physical / graph neural network-based approximation to the widely popular AM1-BCC charge model that is orders of magnitude faster while maintaining accuracy comparable to differences in AM1-BCC implementations. Our hybrid approach couples a graph neural network to a streamlined charge equilibration approach in order to predict molecule-specific atomic electronegativity and hardness parameters, followed by analytical determination of optimal charge-equilibrated parameters that preserves total molecular charge. This hybrid approach scales linearly with the number of atoms, enabling, for the first time, the use of fully consistent charge models for small molecules and biopolymers for the construction of next-generation self-consistent biomolecular force fields. Implemented in the free and open source package \texttt{espaloma\_charge}, this approach provides drop-in replacements for both AmberTools \texttt{antechamber} and the Open Force Field Toolkit charging workflows, in addition to stand-alone charge generation interfaces. Source code is available at \url{https://github.com/choderalab/espaloma_charge}.
Spatial Attention Kinetic Networks with E(n)-Equivariance
Jan 24, 2023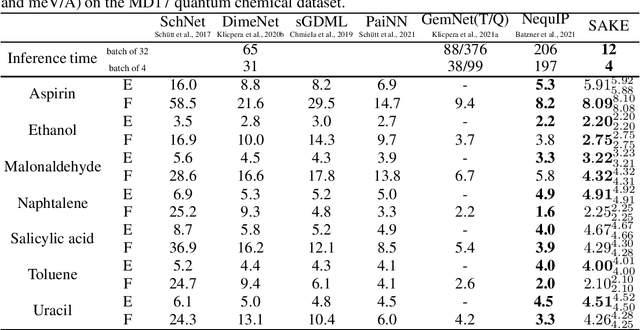
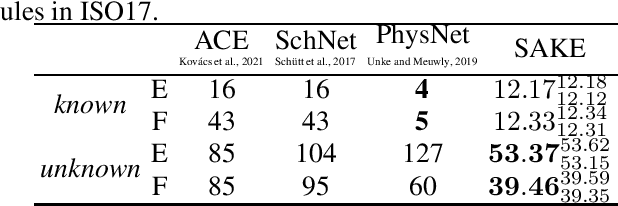
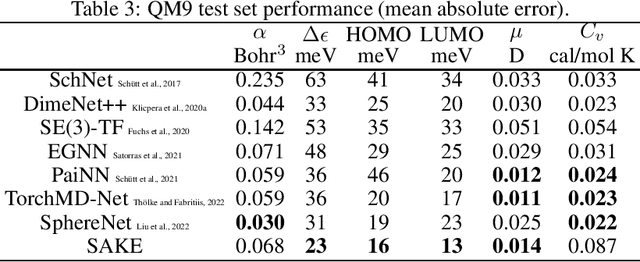
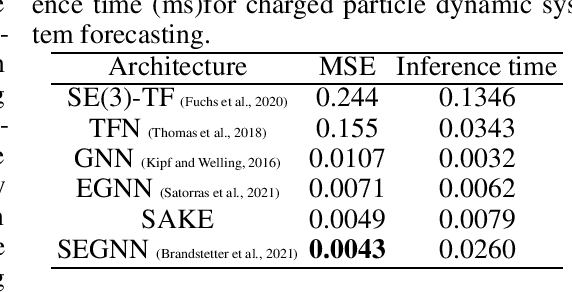
Neural networks that are equivariant to rotations, translations, reflections, and permutations on n-dimensional geometric space have shown promise in physical modeling for tasks such as accurately but inexpensively modeling complex potential energy surfaces to guiding the sampling of complex dynamical systems or forecasting their time evolution. Current state-of-the-art methods employ spherical harmonics to encode higher-order interactions among particles, which are computationally expensive. In this paper, we propose a simple alternative functional form that uses neurally parametrized linear combinations of edge vectors to achieve equivariance while still universally approximating node environments. Incorporating this insight, we design spatial attention kinetic networks with E(n)-equivariance, or SAKE, which are competitive in many-body system modeling tasks while being significantly faster.
SPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials
Sep 21, 2022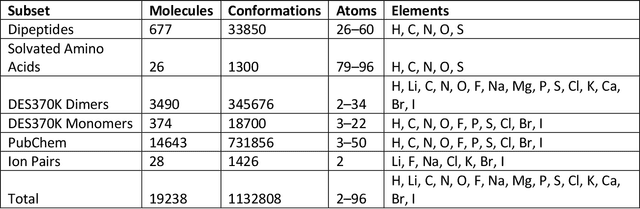
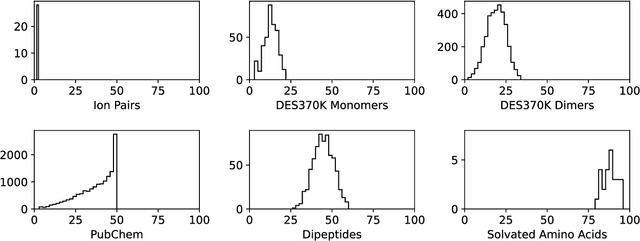

Machine learning potentials are an important tool for molecular simulation, but their development is held back by a shortage of high quality datasets to train them on. We describe the SPICE dataset, a new quantum chemistry dataset for training potentials relevant to simulating drug-like small molecules interacting with proteins. It contains over 1.1 million conformations for a diverse set of small molecules, dimers, dipeptides, and solvated amino acids. It includes 15 elements, charged and uncharged molecules, and a wide range of covalent and non-covalent interactions. It provides both forces and energies calculated at the {\omega}B97M-D3(BJ)/def2-TZVPPD level of theory, along with other useful quantities such as multipole moments and bond orders. We train a set of machine learning potentials on it and demonstrate that they can achieve chemical accuracy across a broad region of chemical space. It can serve as a valuable resource for the creation of transferable, ready to use potential functions for use in molecular simulations.
NNP/MM: Fast molecular dynamics simulations with machine learning potentials and molecular mechanics
Jan 20, 2022
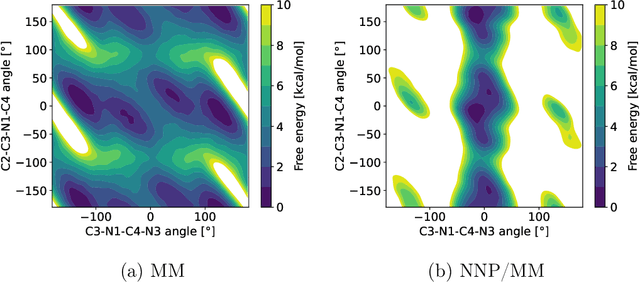
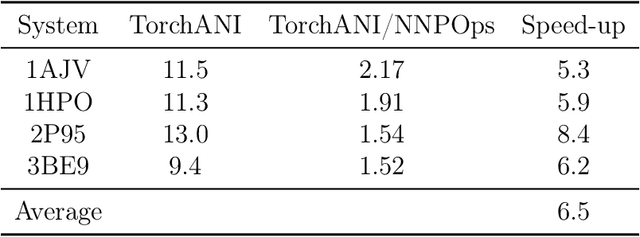
Parametric and non-parametric machine learning potentials have emerged recently as a way to improve the accuracy of bio-molecular simulations. Here, we present NNP/MM, an hybrid method integrating neural network potentials (NNPs) and molecular mechanics (MM). It allows to simulate a part of molecular system with NNP, while the rest is simulated with MM for efficiency. The method is currently available in ACEMD using OpenMM plugins to optimize the performance of NNPs. The achieved performance is slower but comparable to the state-of-the-art GPU-accelerated MM simulations. We validated NNP/MM by performing MD simulations of four protein-ligand complexes, where NNP is used for the intra-molecular interactions of a lignad and MM for the rest interactions. This shows that NNP can already replace MM for small molecules in protein-ligand simulations. The combined sampling of each complex is 1 microsecond, which are the longest simulations of NNP/MM ever reported. Finally, we have made the setup of the NNP/MM simulations simple and user-friendly.
End-to-End Differentiable Molecular Mechanics Force Field Construction
Oct 02, 2020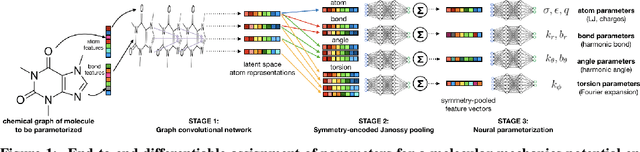
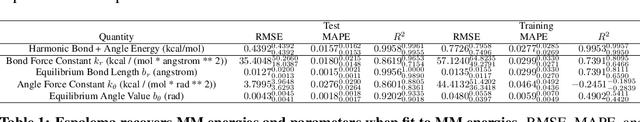
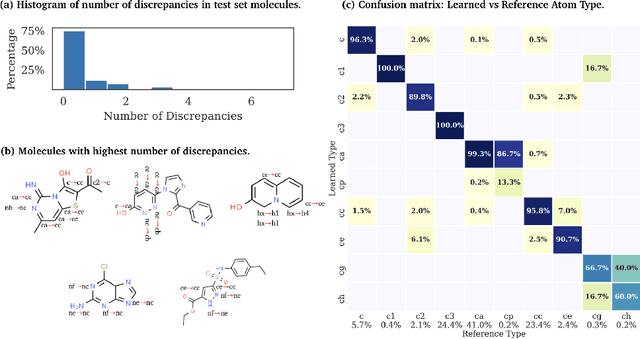

Molecular mechanics (MM) potentials have long been a workhorse of computational chemistry. Leveraging accuracy and speed, these functional forms find use in a wide variety of applications from rapid virtual screening to detailed free energy calculations. Traditionally, MM potentials have relied on human-curated, inflexible, and poorly extensible discrete chemical perception rules (atom types) for applying parameters to molecules or biopolymers, making them difficult to optimize to fit quantum chemical or physical property data. Here, we propose an alternative approach that uses graph nets to perceive chemical environments, producing continuous atom embeddings from which valence and nonbonded parameters can be predicted using a feed-forward neural network. Since all stages are built using smooth functions, the entire process of chemical perception and parameter assignment is differentiable end-to-end with respect to model parameters, allowing new force fields to be easily constructed, extended, and applied to arbitrary molecules. We show that this approach has the capacity to reproduce legacy atom types and can be fit to MM and QM energies and forces, among other targets.