 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnd-to-End Differentiable Molecular Mechanics Force Field Construction
Oct 02, 2020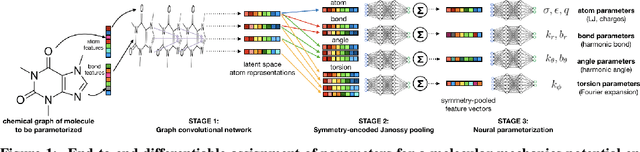
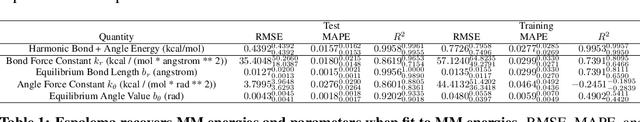
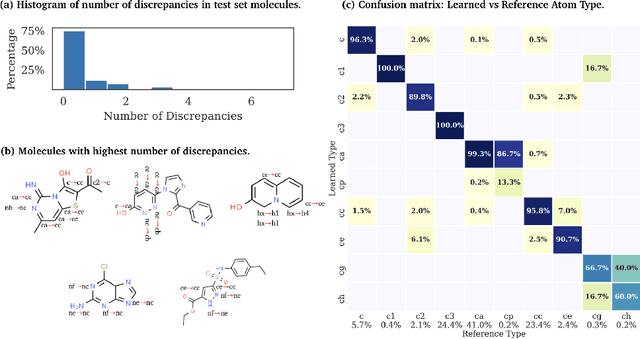

Molecular mechanics (MM) potentials have long been a workhorse of computational chemistry. Leveraging accuracy and speed, these functional forms find use in a wide variety of applications from rapid virtual screening to detailed free energy calculations. Traditionally, MM potentials have relied on human-curated, inflexible, and poorly extensible discrete chemical perception rules (atom types) for applying parameters to molecules or biopolymers, making them difficult to optimize to fit quantum chemical or physical property data. Here, we propose an alternative approach that uses graph nets to perceive chemical environments, producing continuous atom embeddings from which valence and nonbonded parameters can be predicted using a feed-forward neural network. Since all stages are built using smooth functions, the entire process of chemical perception and parameter assignment is differentiable end-to-end with respect to model parameters, allowing new force fields to be easily constructed, extended, and applied to arbitrary molecules. We show that this approach has the capacity to reproduce legacy atom types and can be fit to MM and QM energies and forces, among other targets.
Graph Nets for Partial Charge Prediction
Sep 17, 2019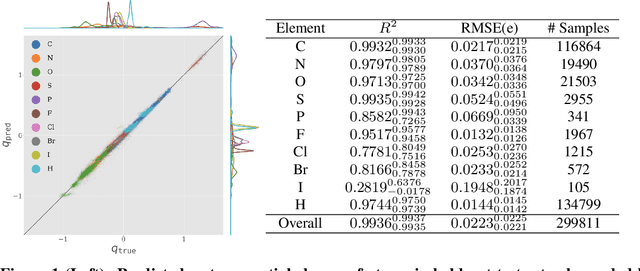
Atomic partial charges are crucial parameters for Molecular Dynamics (MD) simulations, molecular mechanics calculations, and virtual screening, as they determine the electrostatic contributions to interaction energies. Current methods for calculating partial charges, however, are either slow and scale poorly with molecular size (quantum chemical methods) or unreliable (empirical methods). Here, we present a new charge derivation method based on Graph Nets---a set of update and aggregate functions that operate on molecular topologies and propagate information thereon---that could approximate charges derived from Density Functional Theory (DFT) calculations with high accuracy and an over 500-fold speed up.