 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials
Paper and Code
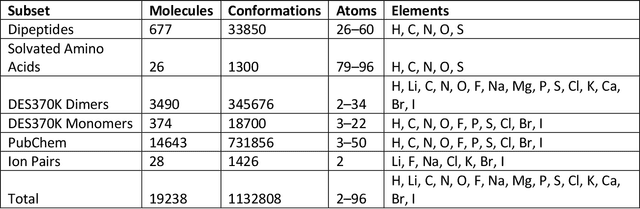
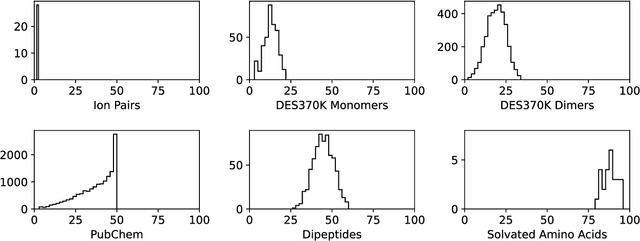

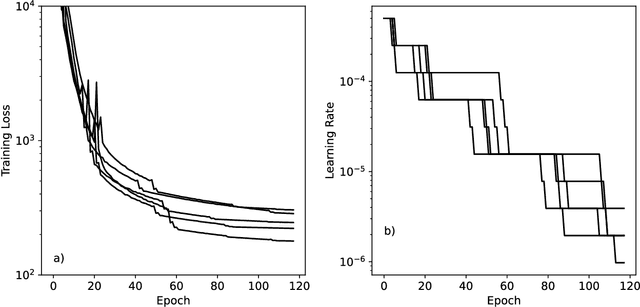
Machine learning potentials are an important tool for molecular simulation, but their development is held back by a shortage of high quality datasets to train them on. We describe the SPICE dataset, a new quantum chemistry dataset for training potentials relevant to simulating drug-like small molecules interacting with proteins. It contains over 1.1 million conformations for a diverse set of small molecules, dimers, dipeptides, and solvated amino acids. It includes 15 elements, charged and uncharged molecules, and a wide range of covalent and non-covalent interactions. It provides both forces and energies calculated at the {\omega}B97M-D3(BJ)/def2-TZVPPD level of theory, along with other useful quantities such as multipole moments and bond orders. We train a set of machine learning potentials on it and demonstrate that they can achieve chemical accuracy across a broad region of chemical space. It can serve as a valuable resource for the creation of transferable, ready to use potential functions for use in molecular simulations.