 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSELFIES and the future of molecular string representations
Mar 31, 2022
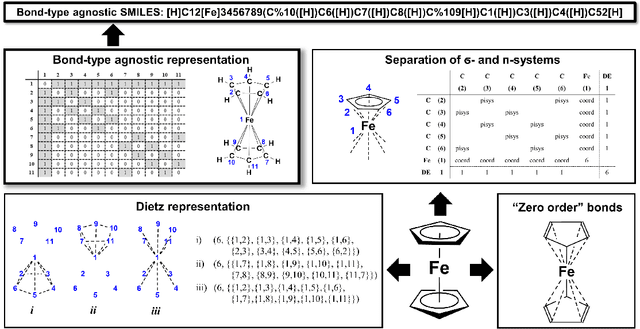
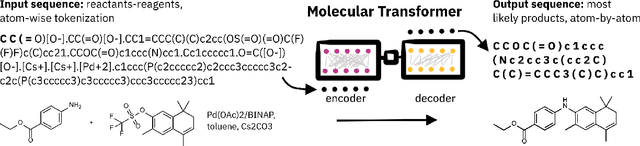
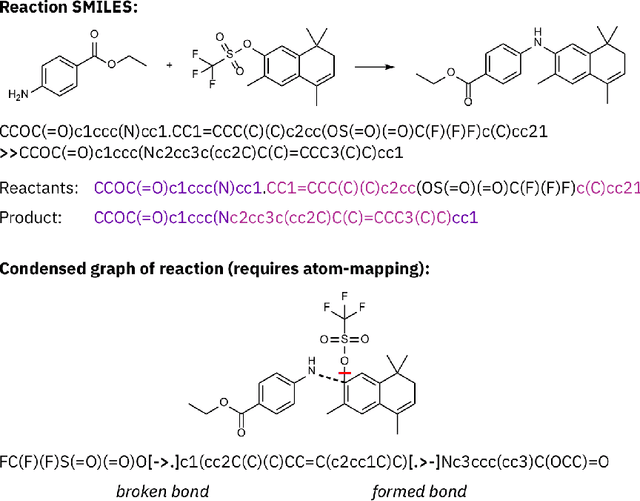
Artificial intelligence (AI) and machine learning (ML) are expanding in popularity for broad applications to challenging tasks in chemistry and materials science. Examples include the prediction of properties, the discovery of new reaction pathways, or the design of new molecules. The machine needs to read and write fluently in a chemical language for each of these tasks. Strings are a common tool to represent molecular graphs, and the most popular molecular string representation, SMILES, has powered cheminformatics since the late 1980s. However, in the context of AI and ML in chemistry, SMILES has several shortcomings -- most pertinently, most combinations of symbols lead to invalid results with no valid chemical interpretation. To overcome this issue, a new language for molecules was introduced in 2020 that guarantees 100\% robustness: SELFIES (SELF-referencIng Embedded Strings). SELFIES has since simplified and enabled numerous new applications in chemistry. In this manuscript, we look to the future and discuss molecular string representations, along with their respective opportunities and challenges. We propose 16 concrete Future Projects for robust molecular representations. These involve the extension toward new chemical domains, exciting questions at the interface of AI and robust languages and interpretability for both humans and machines. We hope that these proposals will inspire several follow-up works exploiting the full potential of molecular string representations for the future of AI in chemistry and materials science.
Coarse Graining Molecular Dynamics with Graph Neural Networks
Aug 21, 2020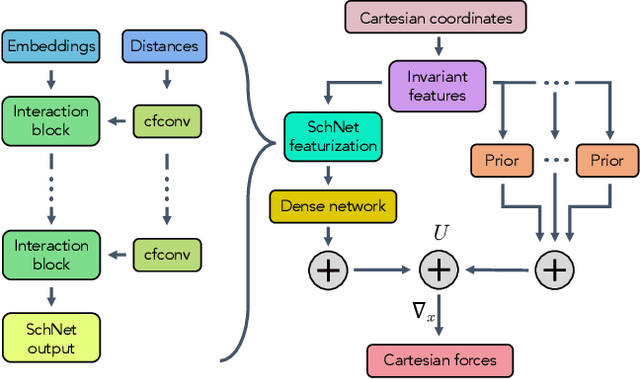
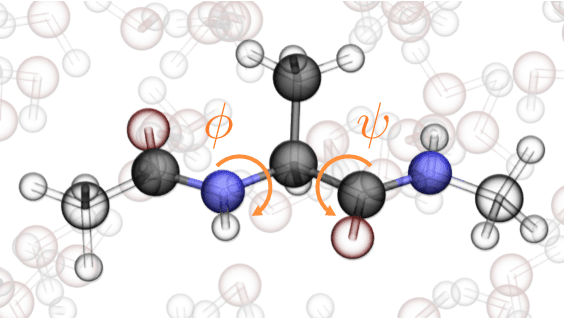
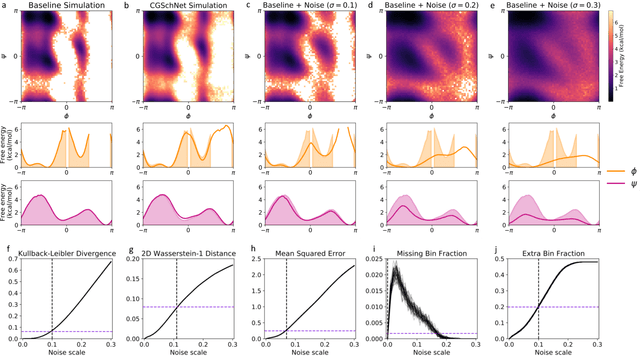
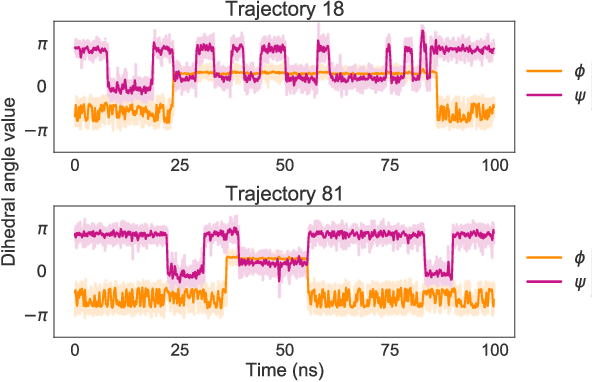
Coarse graining enables the investigation of molecular dynamics for larger systems and at longer timescales than is possible at atomic resolution. However, a coarse graining model must be formulated such that the conclusions we draw from it are consistent with the conclusions we would draw from a model at a finer level of detail. It has been proven that a force matching scheme defines a thermodynamically consistent coarse-grained model for an atomistic system in the variational limit. Wang et al. [ACS Cent. Sci. 5, 755 (2019)] demonstrated that the existence of such a variational limit enables the use of a supervised machine learning framework to generate a coarse-grained force field, which can then be used for simulation in the coarse-grained space. Their framework, however, requires the manual input of molecular features upon which to machine learn the force field. In the present contribution, we build upon the advance of Wang et al.and introduce a hybrid architecture for the machine learning of coarse-grained force fields that learns their own features via a subnetwork that leverages continuous filter convolutions on a graph neural network architecture. We demonstrate that this framework succeeds at reproducing the thermodynamics for small biomolecular systems. Since the learned molecular representations are inherently transferable, the architecture presented here sets the stage for the development of machine-learned, coarse-grained force fields that are transferable across molecular systems.