 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImplicit Transfer Operator Learning: Multiple Time-Resolution Surrogates for Molecular Dynamics
May 29, 2023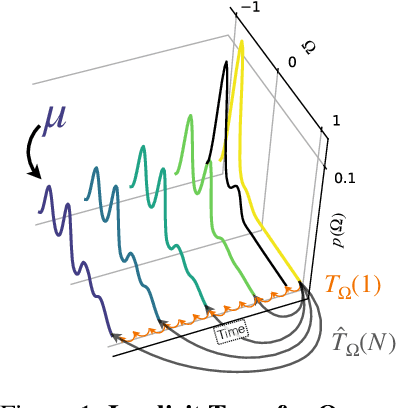

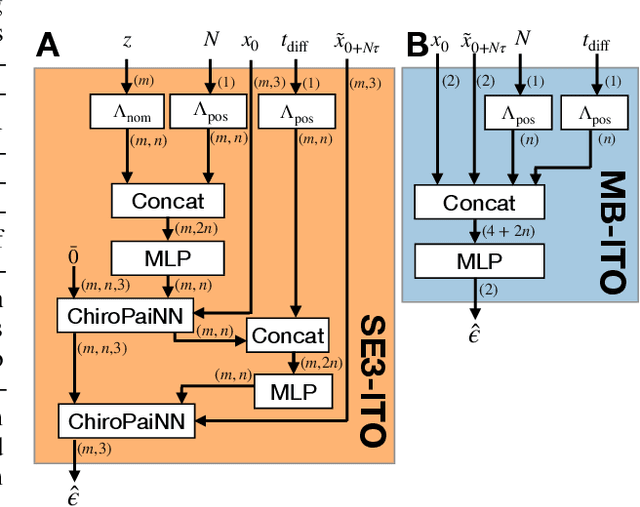

Computing properties of molecular systems rely on estimating expectations of the (unnormalized) Boltzmann distribution. Molecular dynamics (MD) is a broadly adopted technique to approximate such quantities. However, stable simulations rely on very small integration time-steps ($10^{-15}\,\mathrm{s}$), whereas convergence of some moments, e.g. binding free energy or rates, might rely on sampling processes on time-scales as long as $10^{-1}\, \mathrm{s}$, and these simulations must be repeated for every molecular system independently. Here, we present Implict Transfer Operator (ITO) Learning, a framework to learn surrogates of the simulation process with multiple time-resolutions. We implement ITO with denoising diffusion probabilistic models with a new SE(3) equivariant architecture and show the resulting models can generate self-consistent stochastic dynamics across multiple time-scales, even when the system is only partially observed. Finally, we present a coarse-grained CG-SE3-ITO model which can quantitatively model all-atom molecular dynamics using only coarse molecular representations. As such, ITO provides an important step towards multiple time- and space-resolution acceleration of MD.
Transition1x -- a Dataset for Building Generalizable Reactive Machine Learning Potentials
Jul 25, 2022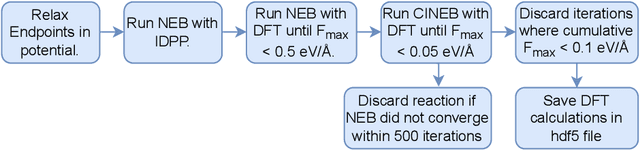
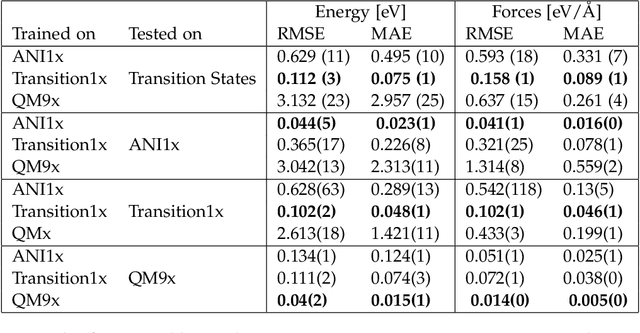
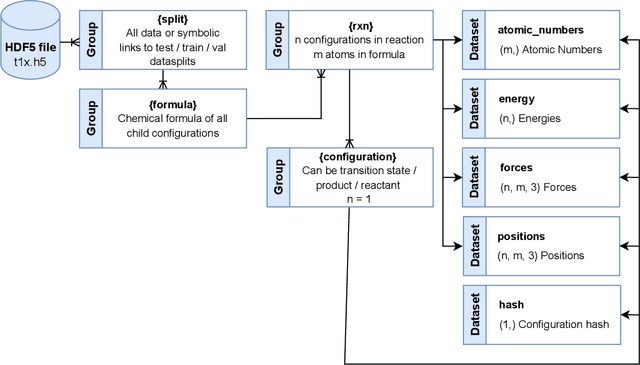
Machine Learning (ML) models have, in contrast to their usefulness in molecular dynamics studies, had limited success as surrogate potentials for reaction barrier search. It is due to the scarcity of training data in relevant transition state regions of chemical space. Currently, available datasets for training ML models on small molecular systems almost exclusively contain configurations at or near equilibrium. In this work, we present the dataset Transition1x containing 9.6 million Density Functional Theory (DFT) calculations of forces and energies of molecular configurations on and around reaction pathways at the wB97x/6-31G(d) level of theory. The data was generated by running Nudged Elastic Band (NEB) calculations with DFT on 10k reactions while saving intermediate calculations. We train state-of-the-art equivariant graph message-passing neural network models on Transition1x and cross-validate on the popular ANI1x and QM9 datasets. We show that ML models cannot learn features in transition-state regions solely by training on hitherto popular benchmark datasets. Transition1x is a new challenging benchmark that will provide an important step towards developing next-generation ML force fields that also work far away from equilibrium configurations and reactive systems.
NeuralNEB -- Neural Networks can find Reaction Paths Fast
Jul 21, 2022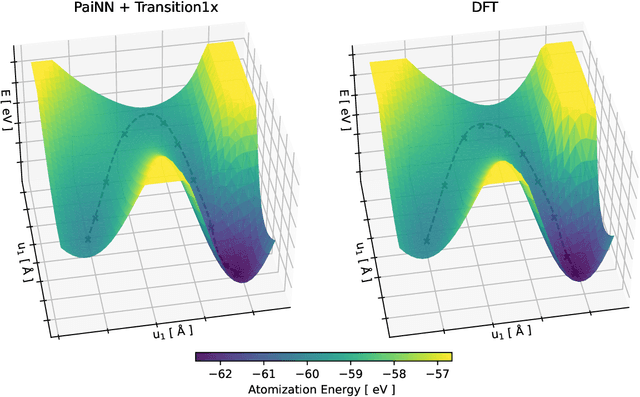
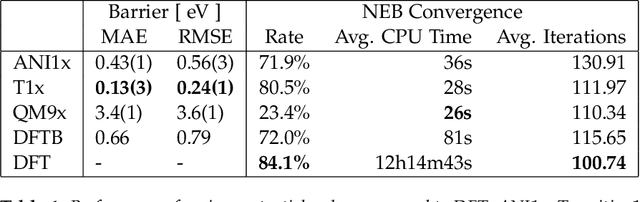
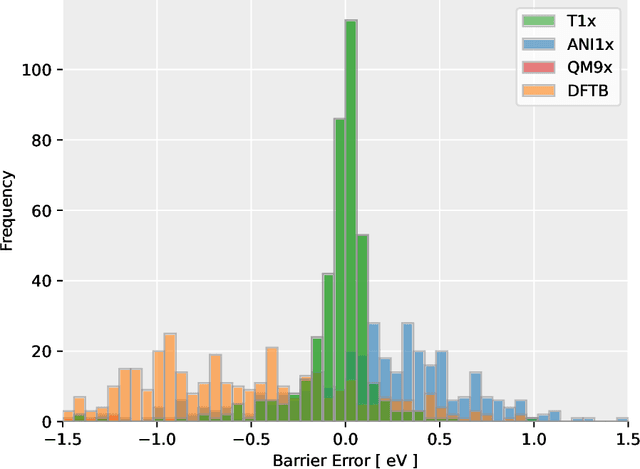
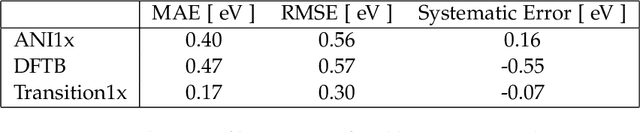
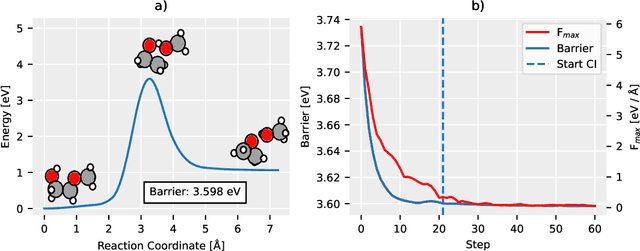
Quantum mechanical methods like Density Functional Theory (DFT) are used with great success alongside efficient search algorithms for studying kinetics of reactive systems. However, DFT is prohibitively expensive for large scale exploration. Machine Learning (ML) models have turned out to be excellent emulators of small molecule DFT calculations and could possibly replace DFT in such tasks. For kinetics, success relies primarily on the models capability to accurately predict the Potential Energy Surface (PES) around transition-states and Minimal Energy Paths (MEPs). Previously this has not been possible due to scarcity of relevant data in the literature. In this paper we train state of the art equivariant Graph Neural Network (GNN)-based models on around 10.000 elementary reactions from the Transition1x dataset. We apply the models as potentials for the Nudged Elastic Band (NEB) algorithm and achieve a Mean Average Error (MAE) of 0.13+/-0.03 eV on barrier energies on unseen reactions. We compare the results against equivalent models trained on QM9 and ANI1x. We also compare with and outperform Density Functional based Tight Binding (DFTB) on both accuracy and computational resource. The implication is that ML models, given relevant data, are now at a level where they can be applied for downstream tasks in quantum chemistry transcending prediction of simple molecular features.