 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCoherent energy and force uncertainty in deep learning force fields
Dec 07, 2023


In machine learning energy potentials for atomic systems, forces are commonly obtained as the negative derivative of the energy function with respect to atomic positions. To quantify aleatoric uncertainty in the predicted energies, a widely used modeling approach involves predicting both a mean and variance for each energy value. However, this model is not differentiable under the usual white noise assumption, so energy uncertainty does not naturally translate to force uncertainty. In this work we propose a machine learning potential energy model in which energy and force aleatoric uncertainty are linked through a spatially correlated noise process. We demonstrate our approach on an equivariant messages passing neural network potential trained on energies and forces on two out-of-equilibrium molecular datasets. Furthermore, we also show how to obtain epistemic uncertainties in this setting based on a Bayesian interpretation of deep ensemble models.
Graph Neural Network Interatomic Potential Ensembles with Calibrated Aleatoric and Epistemic Uncertainty on Energy and Forces
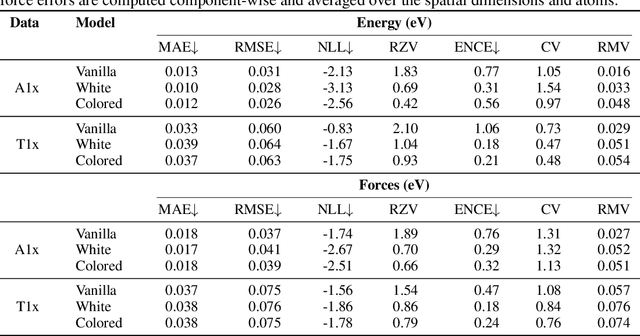
May 10, 2023Inexpensive machine learning potentials are increasingly being used to speed up structural optimization and molecular dynamics simulations of materials by iteratively predicting and applying interatomic forces. In these settings, it is crucial to detect when predictions are unreliable to avoid wrong or misleading results. Here, we present a complete framework for training and recalibrating graph neural network ensemble models to produce accurate predictions of energy and forces with calibrated uncertainty estimates. The proposed method considers both epistemic and aleatoric uncertainty and the total uncertainties are recalibrated post hoc using a nonlinear scaling function to achieve good calibration on previously unseen data, without loss of predictive accuracy. The method is demonstrated and evaluated on two challenging, publicly available datasets, ANI-1x (Smith et al.) and Transition1x (Schreiner et al.), both containing diverse conformations far from equilibrium. A detailed analysis of the predictive performance and uncertainty calibration is provided. In all experiments, the proposed method achieved low prediction error and good uncertainty calibration, with predicted uncertainty correlating with expected error, on energy and forces. To the best of our knowledge, the method presented in this paper is the first to consider a complete framework for obtaining calibrated epistemic and aleatoric uncertainty predictions on both energy and forces in ML potentials.
Transition1x -- a Dataset for Building Generalizable Reactive Machine Learning Potentials
Jul 25, 2022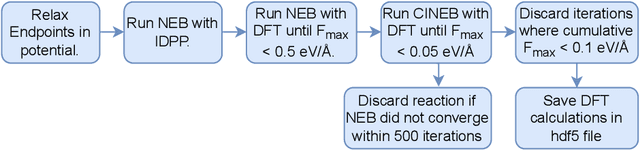
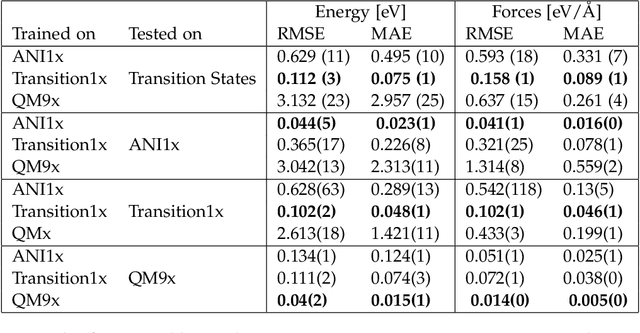
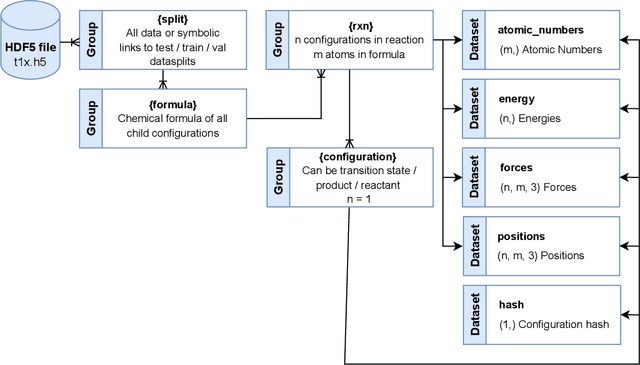
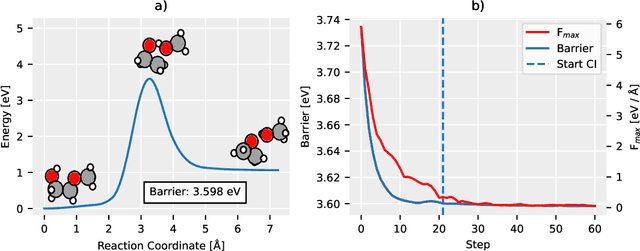
Machine Learning (ML) models have, in contrast to their usefulness in molecular dynamics studies, had limited success as surrogate potentials for reaction barrier search. It is due to the scarcity of training data in relevant transition state regions of chemical space. Currently, available datasets for training ML models on small molecular systems almost exclusively contain configurations at or near equilibrium. In this work, we present the dataset Transition1x containing 9.6 million Density Functional Theory (DFT) calculations of forces and energies of molecular configurations on and around reaction pathways at the wB97x/6-31G(d) level of theory. The data was generated by running Nudged Elastic Band (NEB) calculations with DFT on 10k reactions while saving intermediate calculations. We train state-of-the-art equivariant graph message-passing neural network models on Transition1x and cross-validate on the popular ANI1x and QM9 datasets. We show that ML models cannot learn features in transition-state regions solely by training on hitherto popular benchmark datasets. Transition1x is a new challenging benchmark that will provide an important step towards developing next-generation ML force fields that also work far away from equilibrium configurations and reactive systems.
Calibrated Uncertainty for Molecular Property Prediction using Ensembles of Message Passing Neural Networks
Jul 13, 2021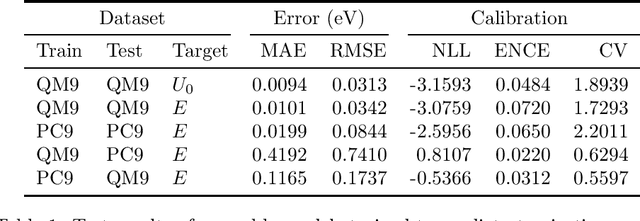

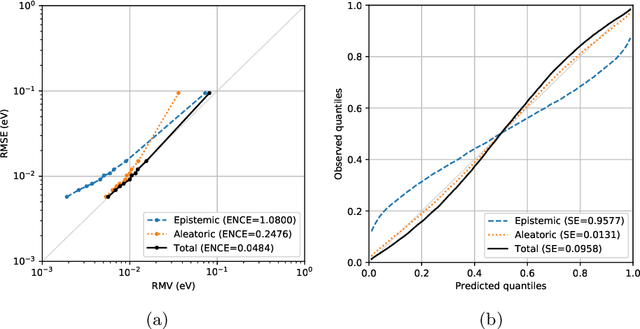
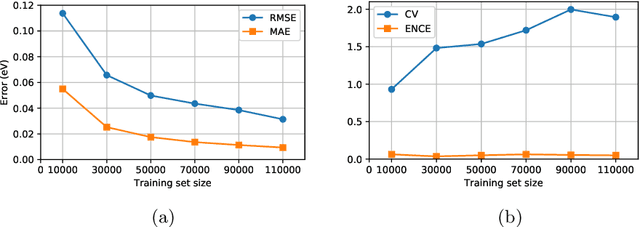
Data-driven methods based on machine learning have the potential to accelerate analysis of atomic structures. However, machine learning models can produce overconfident predictions and it is therefore crucial to detect and handle uncertainty carefully. Here, we extend a message passing neural network designed specifically for predicting properties of molecules and materials with a calibrated probabilistic predictive distribution. The method presented in this paper differs from the previous work by considering both aleatoric and epistemic uncertainty in a unified framework, and by re-calibrating the predictive distribution on unseen data. Through computer experiments, we show that our approach results in accurate models for predicting molecular formation energies with calibrated uncertainty in and out of the training data distribution on two public molecular benchmark datasets, QM9 and PC9. The proposed method provides a general framework for training and evaluating neural network ensemble models that are able to produce accurate predictions of properties of molecules with calibrated uncertainty.