 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCoherent energy and force uncertainty in deep learning force fields
Dec 07, 2023
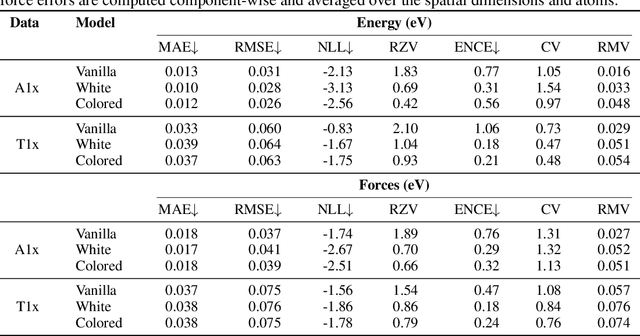


In machine learning energy potentials for atomic systems, forces are commonly obtained as the negative derivative of the energy function with respect to atomic positions. To quantify aleatoric uncertainty in the predicted energies, a widely used modeling approach involves predicting both a mean and variance for each energy value. However, this model is not differentiable under the usual white noise assumption, so energy uncertainty does not naturally translate to force uncertainty. In this work we propose a machine learning potential energy model in which energy and force aleatoric uncertainty are linked through a spatially correlated noise process. We demonstrate our approach on an equivariant messages passing neural network potential trained on energies and forces on two out-of-equilibrium molecular datasets. Furthermore, we also show how to obtain epistemic uncertainties in this setting based on a Bayesian interpretation of deep ensemble models.
Graph Neural Network Interatomic Potential Ensembles with Calibrated Aleatoric and Epistemic Uncertainty on Energy and Forces
May 10, 2023Inexpensive machine learning potentials are increasingly being used to speed up structural optimization and molecular dynamics simulations of materials by iteratively predicting and applying interatomic forces. In these settings, it is crucial to detect when predictions are unreliable to avoid wrong or misleading results. Here, we present a complete framework for training and recalibrating graph neural network ensemble models to produce accurate predictions of energy and forces with calibrated uncertainty estimates. The proposed method considers both epistemic and aleatoric uncertainty and the total uncertainties are recalibrated post hoc using a nonlinear scaling function to achieve good calibration on previously unseen data, without loss of predictive accuracy. The method is demonstrated and evaluated on two challenging, publicly available datasets, ANI-1x (Smith et al.) and Transition1x (Schreiner et al.), both containing diverse conformations far from equilibrium. A detailed analysis of the predictive performance and uncertainty calibration is provided. In all experiments, the proposed method achieved low prediction error and good uncertainty calibration, with predicted uncertainty correlating with expected error, on energy and forces. To the best of our knowledge, the method presented in this paper is the first to consider a complete framework for obtaining calibrated epistemic and aleatoric uncertainty predictions on both energy and forces in ML potentials.
Graph neural networks for fast electron density estimation of molecules, liquids, and solids
Dec 01, 2021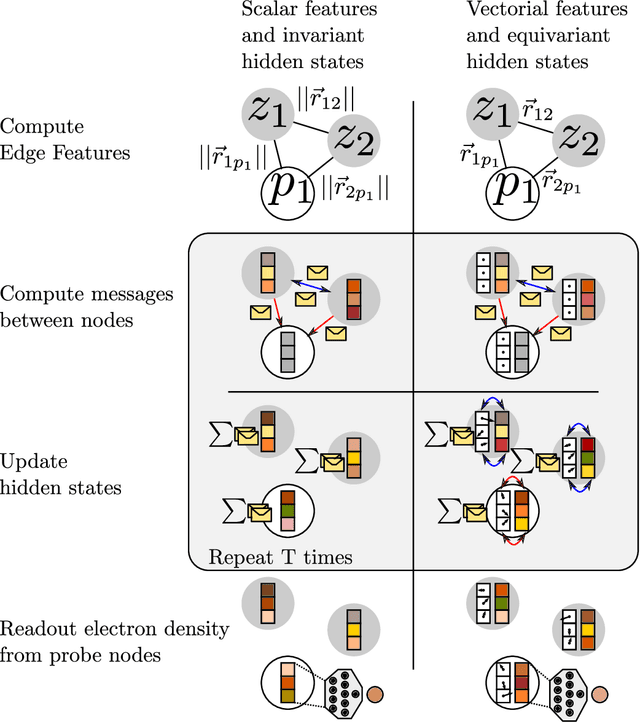
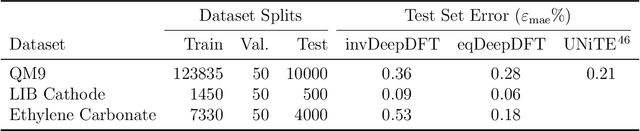
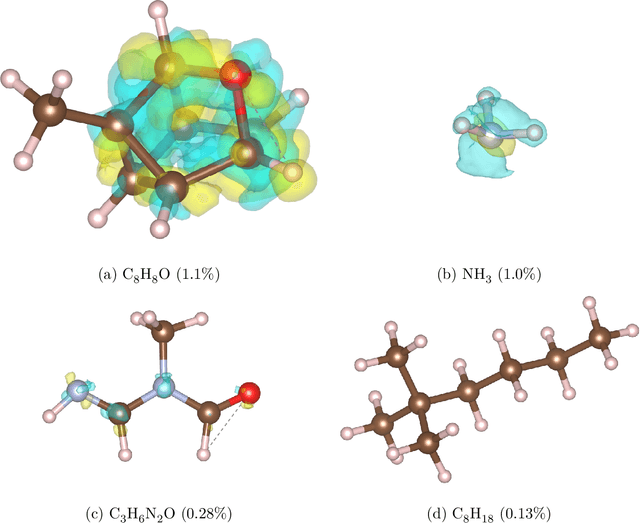
Electron density $\rho(\vec{r})$ is the fundamental variable in the calculation of ground state energy with density functional theory (DFT). Beyond total energy, features in $\rho(\vec{r})$ distribution and modifications in $\rho(\vec{r})$ are often used to capture critical physicochemical phenomena in functional materials and molecules at the electronic scale. Methods providing access to $\rho(\vec{r})$ of complex disordered systems with little computational cost can be a game changer in the expedited exploration of materials phase space towards the inverse design of new materials with better functionalities. We present a machine learning framework for the prediction of $\rho(\vec{r})$. The model is based on equivariant graph neural networks and the electron density is predicted at special query point vertices that are part of the message passing graph, but only receive messages. The model is tested across multiple data sets of molecules (QM9), liquid ethylene carbonate electrolyte (EC) and LixNiyMnzCo(1-y-z)O2 lithium ion battery cathodes (NMC). For QM9 molecules, the accuracy of the proposed model exceeds typical variability in $\rho(\vec{r})$ obtained from DFT done with different exchange-correlation functional and show beyond the state of the art accuracy. The accuracy is even better for the mixed oxide (NMC) and electrolyte (EC) datasets. The linear scaling model's capacity to probe thousands of points simultaneously permits calculation of $\rho(\vec{r})$ for large complex systems many orders of magnitude faster than DFT allowing screening of disordered functional materials.
Calibrated Uncertainty for Molecular Property Prediction using Ensembles of Message Passing Neural Networks
Jul 13, 2021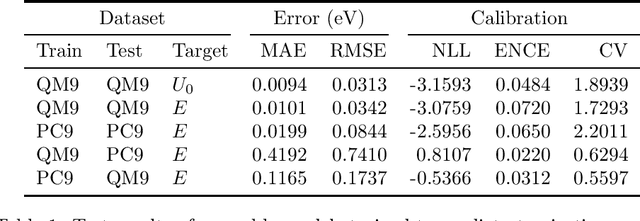

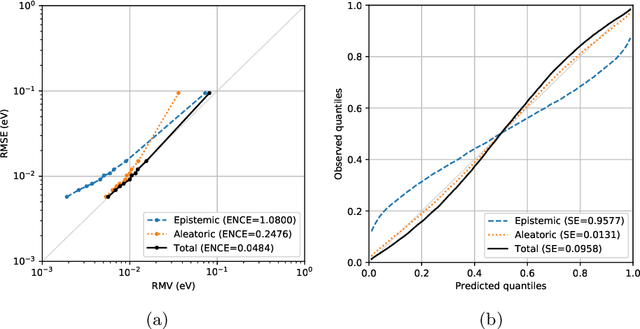
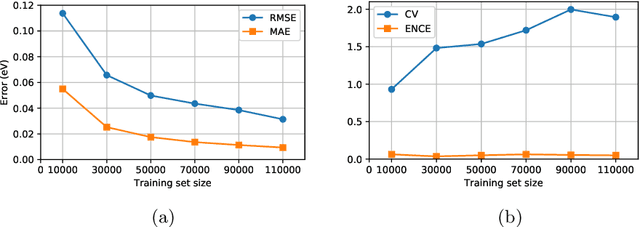
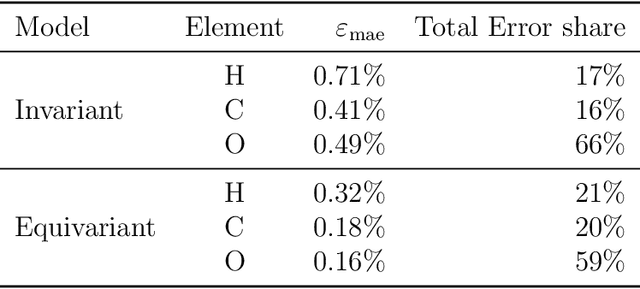
Data-driven methods based on machine learning have the potential to accelerate analysis of atomic structures. However, machine learning models can produce overconfident predictions and it is therefore crucial to detect and handle uncertainty carefully. Here, we extend a message passing neural network designed specifically for predicting properties of molecules and materials with a calibrated probabilistic predictive distribution. The method presented in this paper differs from the previous work by considering both aleatoric and epistemic uncertainty in a unified framework, and by re-calibrating the predictive distribution on unseen data. Through computer experiments, we show that our approach results in accurate models for predicting molecular formation energies with calibrated uncertainty in and out of the training data distribution on two public molecular benchmark datasets, QM9 and PC9. The proposed method provides a general framework for training and evaluating neural network ensemble models that are able to produce accurate predictions of properties of molecules with calibrated uncertainty.
DeepDFT: Neural Message Passing Network for Accurate Charge Density Prediction
Nov 04, 2020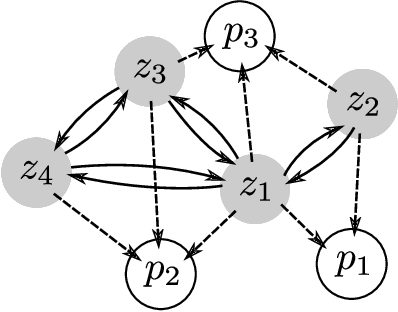
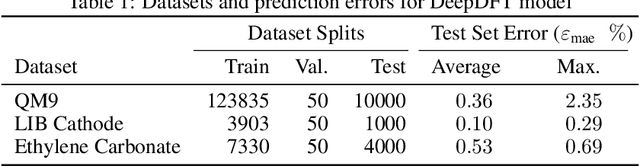
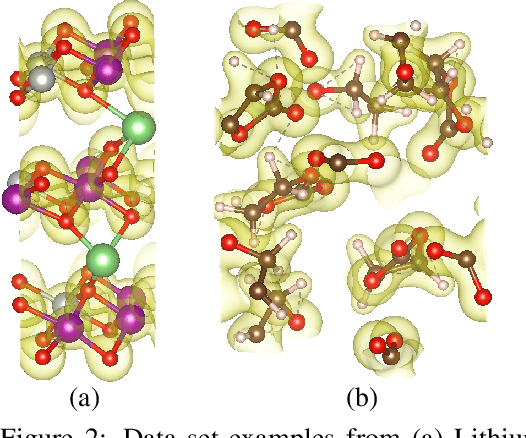
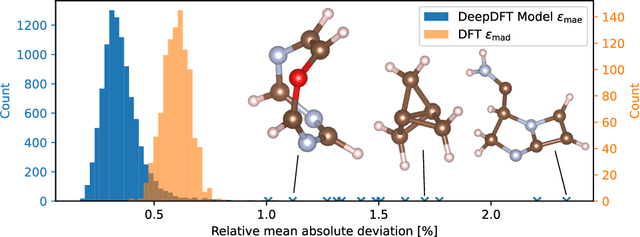
We introduce DeepDFT, a deep learning model for predicting the electronic charge density around atoms, the fundamental variable in electronic structure simulations from which all ground state properties can be calculated. The model is formulated as neural message passing on a graph, consisting of interacting atom vertices and special query point vertices for which the charge density is predicted. The accuracy and scalability of the model are demonstrated for molecules, solids and liquids. The trained model achieves lower average prediction errors than the observed variations in charge density obtained from density functional theory simulations using different exchange correlation functionals.
Materials property prediction using symmetry-labeled graphs as atomic-position independent descriptors
May 16, 2019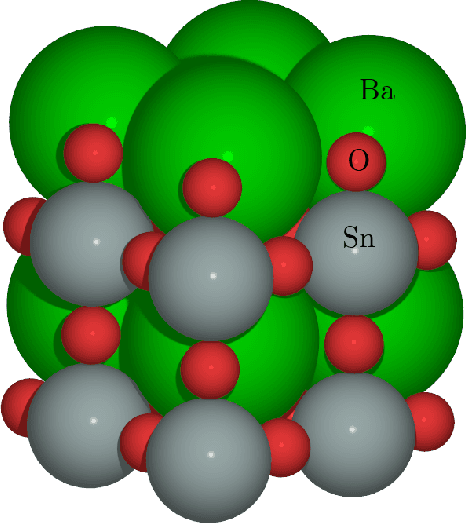
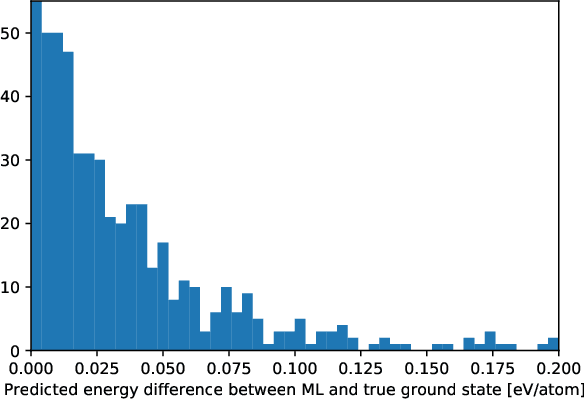
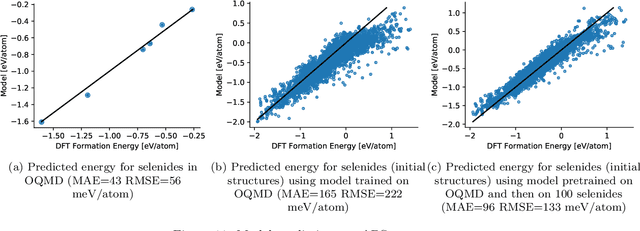

Computational materials screening studies require fast calculation of the properties of thousands of materials. The calculations are often performed with Density Functional Theory (DFT), but the necessary computer time sets limitations for the investigated material space. Therefore, the development of machine learning models for prediction of DFT calculated properties are currently of interest. A particular challenge for \emph{new} materials is that the atomic positions are generally not known. We present a machine learning model for the prediction of DFT-calculated formation energies based on Voronoi quotient graphs and local symmetry classification without the need for detailed information about atomic positions. The model is implemented as a message passing neural network and tested on the Open Quantum Materials Database (OQMD) and the Materials Project database. The test mean absolute error is 20 meV on the OQMD database and 40 meV on Materials Project Database. The possibilities for prediction in a realistic computational screening setting is investigated on a dataset of 5976 ABSe$_3$ selenides with very limited overlap with the OQMD training set. Pretraining on OQMD and subsequent training on 100 selenides result in a mean absolute error below 0.1 eV for the formation energy of the selenides.
Neural Message Passing with Edge Updates for Predicting Properties of Molecules and Materials
Jun 08, 2018

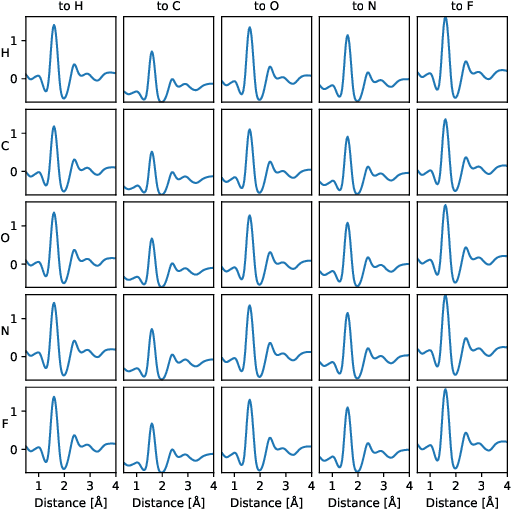

Neural message passing on molecular graphs is one of the most promising methods for predicting formation energy and other properties of molecules and materials. In this work we extend the neural message passing model with an edge update network which allows the information exchanged between atoms to depend on the hidden state of the receiving atom. We benchmark the proposed model on three publicly available datasets (QM9, The Materials Project and OQMD) and show that the proposed model yields superior prediction of formation energies and other properties on all three datasets in comparison with the best published results. Furthermore we investigate different methods for constructing the graph used to represent crystalline structures and we find that using a graph based on K-nearest neighbors achieves better prediction accuracy than using maximum distance cutoff or the Voronoi tessellation graph.