 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBayesian optimization of atomic structures with prior probabilities from universal interatomic potentials
Aug 28, 2024

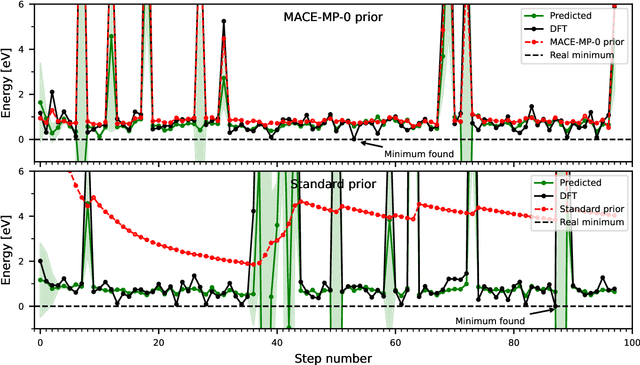
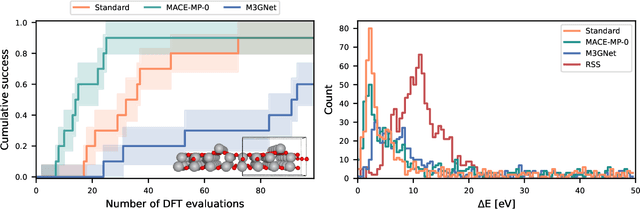
The optimization of atomic structures plays a pivotal role in understanding and designing materials with desired properties. However, conventional methods often struggle with the formidable task of navigating the vast potential energy surface, especially in high-dimensional spaces with numerous local minima. Recent advancements in machine learning-driven surrogate models offer a promising avenue for alleviating this computational burden. In this study, we propose a novel approach that combines the strengths of universal machine learning potentials with a Bayesian approach of the GOFEE/BEACON framework. By leveraging the comprehensive chemical knowledge encoded in pretrained universal machine learning potentials as a prior estimate of energy and forces, we enable the Gaussian process to focus solely on capturing the intricate nuances of the potential energy surface. We demonstrate the efficacy of our approach through comparative analyses across diverse systems, including periodic bulk materials, surface structures, and a cluster.
Materials property prediction using symmetry-labeled graphs as atomic-position independent descriptors
May 16, 2019
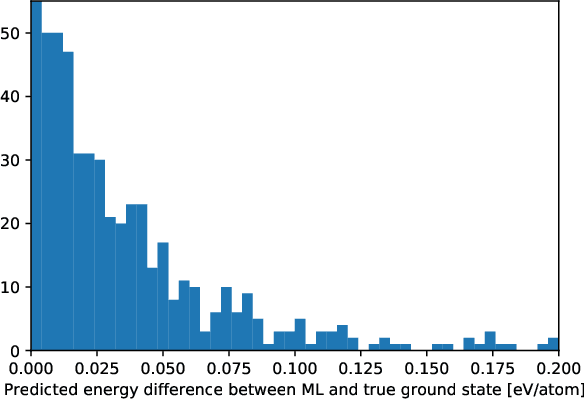
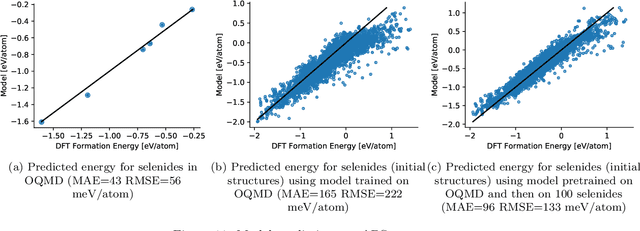

Computational materials screening studies require fast calculation of the properties of thousands of materials. The calculations are often performed with Density Functional Theory (DFT), but the necessary computer time sets limitations for the investigated material space. Therefore, the development of machine learning models for prediction of DFT calculated properties are currently of interest. A particular challenge for \emph{new} materials is that the atomic positions are generally not known. We present a machine learning model for the prediction of DFT-calculated formation energies based on Voronoi quotient graphs and local symmetry classification without the need for detailed information about atomic positions. The model is implemented as a message passing neural network and tested on the Open Quantum Materials Database (OQMD) and the Materials Project database. The test mean absolute error is 20 meV on the OQMD database and 40 meV on Materials Project Database. The possibilities for prediction in a realistic computational screening setting is investigated on a dataset of 5976 ABSe$_3$ selenides with very limited overlap with the OQMD training set. Pretraining on OQMD and subsequent training on 100 selenides result in a mean absolute error below 0.1 eV for the formation energy of the selenides.
Neural Message Passing with Edge Updates for Predicting Properties of Molecules and Materials
Jun 08, 2018

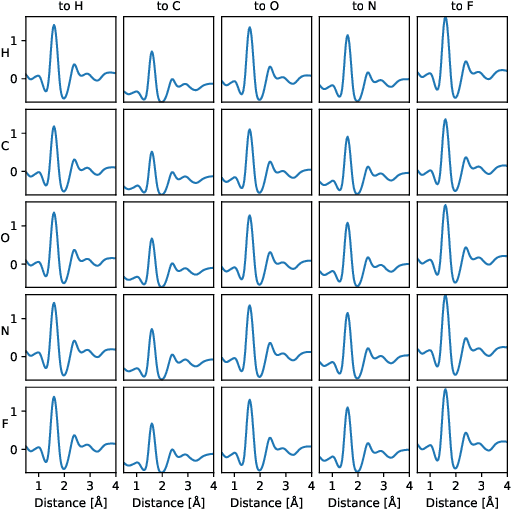

Neural message passing on molecular graphs is one of the most promising methods for predicting formation energy and other properties of molecules and materials. In this work we extend the neural message passing model with an edge update network which allows the information exchanged between atoms to depend on the hidden state of the receiving atom. We benchmark the proposed model on three publicly available datasets (QM9, The Materials Project and OQMD) and show that the proposed model yields superior prediction of formation energies and other properties on all three datasets in comparison with the best published results. Furthermore we investigate different methods for constructing the graph used to represent crystalline structures and we find that using a graph based on K-nearest neighbors achieves better prediction accuracy than using maximum distance cutoff or the Voronoi tessellation graph.