 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMaterials property prediction using symmetry-labeled graphs as atomic-position independent descriptors
Paper and Code
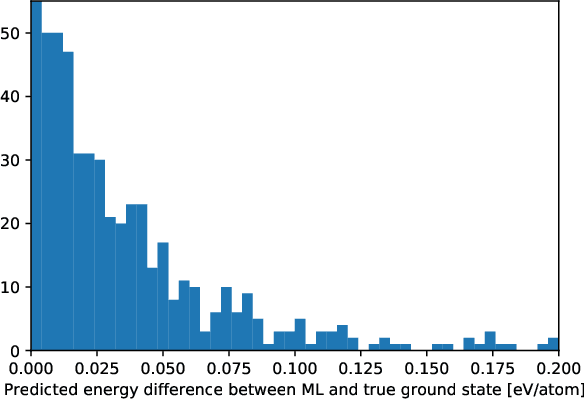
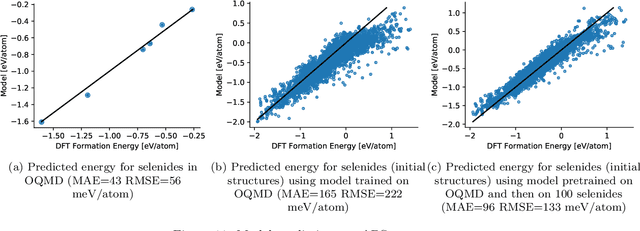

Computational materials screening studies require fast calculation of the properties of thousands of materials. The calculations are often performed with Density Functional Theory (DFT), but the necessary computer time sets limitations for the investigated material space. Therefore, the development of machine learning models for prediction of DFT calculated properties are currently of interest. A particular challenge for \emph{new} materials is that the atomic positions are generally not known. We present a machine learning model for the prediction of DFT-calculated formation energies based on Voronoi quotient graphs and local symmetry classification without the need for detailed information about atomic positions. The model is implemented as a message passing neural network and tested on the Open Quantum Materials Database (OQMD) and the Materials Project database. The test mean absolute error is 20 meV on the OQMD database and 40 meV on Materials Project Database. The possibilities for prediction in a realistic computational screening setting is investigated on a dataset of 5976 ABSe$_3$ selenides with very limited overlap with the OQMD training set. Pretraining on OQMD and subsequent training on 100 selenides result in a mean absolute error below 0.1 eV for the formation energy of the selenides.