 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCalibrated Uncertainty for Molecular Property Prediction using Ensembles of Message Passing Neural Networks
Paper and Code
Jul 13, 2021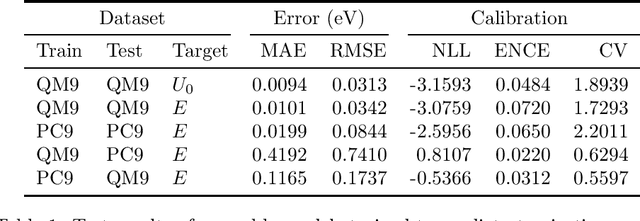

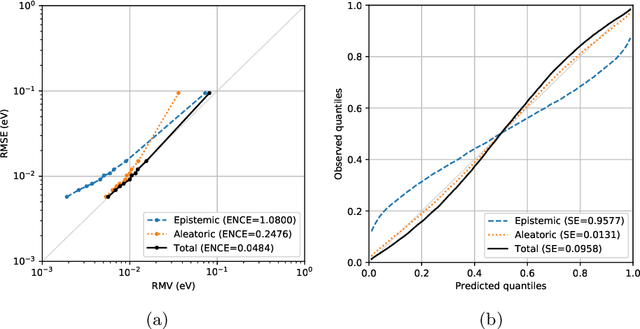
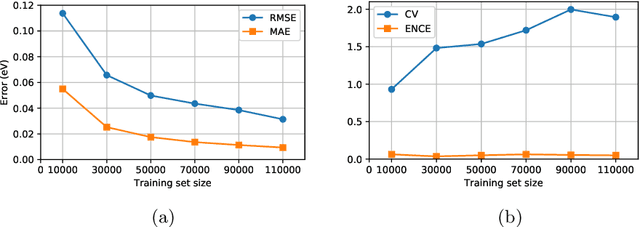
Data-driven methods based on machine learning have the potential to accelerate analysis of atomic structures. However, machine learning models can produce overconfident predictions and it is therefore crucial to detect and handle uncertainty carefully. Here, we extend a message passing neural network designed specifically for predicting properties of molecules and materials with a calibrated probabilistic predictive distribution. The method presented in this paper differs from the previous work by considering both aleatoric and epistemic uncertainty in a unified framework, and by re-calibrating the predictive distribution on unseen data. Through computer experiments, we show that our approach results in accurate models for predicting molecular formation energies with calibrated uncertainty in and out of the training data distribution on two public molecular benchmark datasets, QM9 and PC9. The proposed method provides a general framework for training and evaluating neural network ensemble models that are able to produce accurate predictions of properties of molecules with calibrated uncertainty.