 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGlobal Plane Waves From Local Gaussians: Periodic Charge Densities in a Blink
Jan 27, 2026We introduce ELECTRAFI, a fast, end-to-end differentiable model for predicting periodic charge densities in crystalline materials. ELECTRAFI constructs anisotropic Gaussians in real space and exploits their closed-form Fourier transforms to analytically evaluate plane-wave coefficients via the Poisson summation formula. This formulation delegates non-local and periodic behavior to analytic transforms, enabling reconstruction of the full periodic charge density with a single inverse FFT. By avoiding explicit real-space grid probing, periodic image summation, and spherical harmonic expansions, ELECTRAFI matches or exceeds state-of-the-art accuracy across periodic benchmarks while being up to $633 \times$ faster than the strongest competing method, reconstructing crystal charge densities in a fraction of a second. When used to initialize DFT calculations, ELECTRAFI reduces total DFT compute cost by up to ~20%, whereas slower charge density models negate savings due to high inference times. Our results show that accuracy and inference cost jointly determine end-to-end DFT speedups, and motivate our focus on efficiency.
Acoustic Classification of Maritime Vessels using Learnable Filterbanks
May 29, 2025
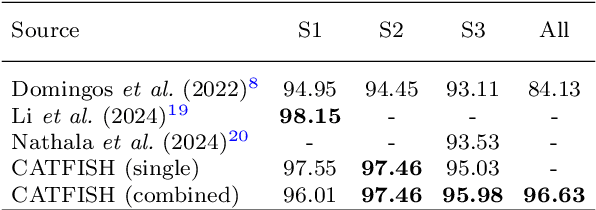

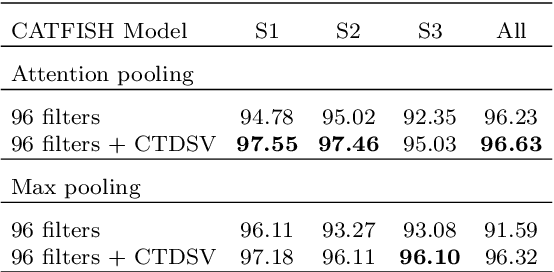
Reliably monitoring and recognizing maritime vessels based on acoustic signatures is complicated by the variability of different recording scenarios. A robust classification framework must be able to generalize across diverse acoustic environments and variable source-sensor distances. To this end, we present a deep learning model with robust performance across different recording scenarios. Using a trainable spectral front-end and temporal feature encoder to learn a Gabor filterbank, the model can dynamically emphasize different frequency components. Trained on the VTUAD hydrophone recordings from the Strait of Georgia, our model, CATFISH, achieves a state-of-the-art 96.63 % percent test accuracy across varying source-sensor distances, surpassing the previous benchmark by over 12 percentage points. We present the model, justify our architectural choices, analyze the learned Gabor filters, and perform ablation studies on sensor data fusion and attention-based pooling.
Autonomous nanoparticle synthesis by design
May 19, 2025Controlled synthesis of materials with specified atomic structures underpins technological advances yet remains reliant on iterative, trial-and-error approaches. Nanoparticles (NPs), whose atomic arrangement dictates their emergent properties, are particularly challenging to synthesise due to numerous tunable parameters. Here, we introduce an autonomous approach explicitly targeting synthesis of atomic-scale structures. Our method autonomously designs synthesis protocols by matching real time experimental total scattering (TS) and pair distribution function (PDF) data to simulated target patterns, without requiring prior synthesis knowledge. We demonstrate this capability at a synchrotron, successfully synthesising two structurally distinct gold NPs: 5 nm decahedral and 10 nm face-centred cubic structures. Ultimately, specifying a simulated target scattering pattern, thus representing a bespoke atomic structure, and obtaining both the synthesised material and its reproducible synthesis protocol on demand may revolutionise materials design. Thus, ScatterLab provides a generalisable blueprint for autonomous, atomic structure-targeted synthesis across diverse systems and applications.
ELECTRA: A Symmetry-breaking Cartesian Network for Charge Density Prediction with Floating Orbitals
Mar 11, 2025We present the Electronic Tensor Reconstruction Algorithm (ELECTRA) - an equivariant model for predicting electronic charge densities using "floating" orbitals. Floating orbitals are a long-standing idea in the quantum chemistry community that promises more compact and accurate representations by placing orbitals freely in space, as opposed to centering all orbitals at the position of atoms. Finding ideal placements of these orbitals requires extensive domain knowledge though, which thus far has prevented widespread adoption. We solve this in a data-driven manner by training a Cartesian tensor network to predict orbital positions along with orbital coefficients. This is made possible through a symmetry-breaking mechanism that is used to learn position displacements with lower symmetry than the input molecule while preserving the rotation equivariance of the charge density itself. Inspired by recent successes of Gaussian Splatting in representing densities in space, we are using Gaussians as our orbitals and predict their weights and covariance matrices. Our method achieves a state-of-the-art balance between computational efficiency and predictive accuracy on established benchmarks.
MolMiner: Transformer architecture for fragment-based autoregressive generation of molecular stories
Nov 10, 2024



Deep generative models for molecular discovery have become a very popular choice in new high-throughput screening paradigms. These models have been developed inheriting from the advances in natural language processing and computer vision, achieving ever greater results. However, generative molecular modelling has unique challenges that are often overlooked. Chemical validity, interpretability of the generation process and flexibility to variable molecular sizes are among some of the remaining challenges for generative models in computational materials design. In this work, we propose an autoregressive approach that decomposes molecular generation into a sequence of discrete and interpretable steps using molecular fragments as units, a 'molecular story'. Enforcing chemical rules in the stories guarantees the chemical validity of the generated molecules, the discrete sequential steps of a molecular story makes the process transparent improving interpretability, and the autoregressive nature of the approach allows the size of the molecule to be a decision of the model. We demonstrate the validity of the approach in a multi-target inverse design of electroactive organic compounds, focusing on the target properties of solubility, redox potential, and synthetic accessibility. Our results show that the model can effectively bias the generation distribution according to the prompted multi-target objective.
Graph Neural Network Interatomic Potential Ensembles with Calibrated Aleatoric and Epistemic Uncertainty on Energy and Forces
May 10, 2023Inexpensive machine learning potentials are increasingly being used to speed up structural optimization and molecular dynamics simulations of materials by iteratively predicting and applying interatomic forces. In these settings, it is crucial to detect when predictions are unreliable to avoid wrong or misleading results. Here, we present a complete framework for training and recalibrating graph neural network ensemble models to produce accurate predictions of energy and forces with calibrated uncertainty estimates. The proposed method considers both epistemic and aleatoric uncertainty and the total uncertainties are recalibrated post hoc using a nonlinear scaling function to achieve good calibration on previously unseen data, without loss of predictive accuracy. The method is demonstrated and evaluated on two challenging, publicly available datasets, ANI-1x (Smith et al.) and Transition1x (Schreiner et al.), both containing diverse conformations far from equilibrium. A detailed analysis of the predictive performance and uncertainty calibration is provided. In all experiments, the proposed method achieved low prediction error and good uncertainty calibration, with predicted uncertainty correlating with expected error, on energy and forces. To the best of our knowledge, the method presented in this paper is the first to consider a complete framework for obtaining calibrated epistemic and aleatoric uncertainty predictions on both energy and forces in ML potentials.
Transition1x -- a Dataset for Building Generalizable Reactive Machine Learning Potentials
Jul 25, 2022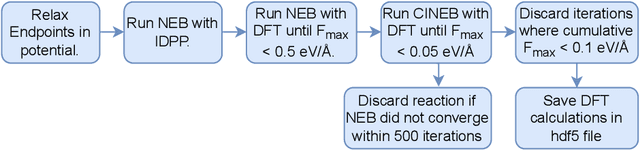
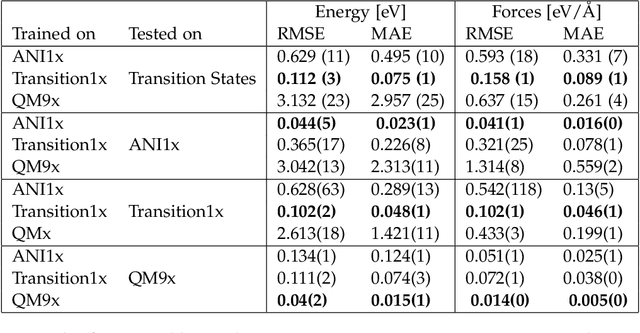
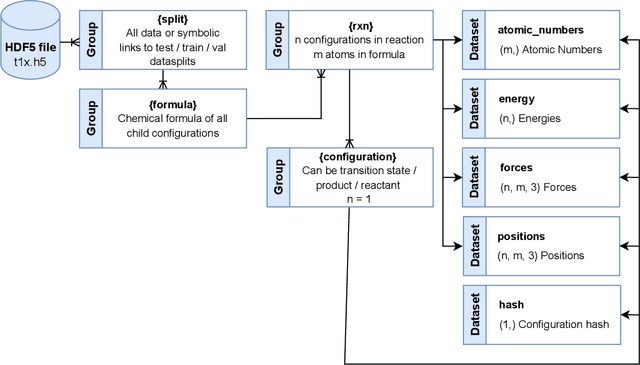
Machine Learning (ML) models have, in contrast to their usefulness in molecular dynamics studies, had limited success as surrogate potentials for reaction barrier search. It is due to the scarcity of training data in relevant transition state regions of chemical space. Currently, available datasets for training ML models on small molecular systems almost exclusively contain configurations at or near equilibrium. In this work, we present the dataset Transition1x containing 9.6 million Density Functional Theory (DFT) calculations of forces and energies of molecular configurations on and around reaction pathways at the wB97x/6-31G(d) level of theory. The data was generated by running Nudged Elastic Band (NEB) calculations with DFT on 10k reactions while saving intermediate calculations. We train state-of-the-art equivariant graph message-passing neural network models on Transition1x and cross-validate on the popular ANI1x and QM9 datasets. We show that ML models cannot learn features in transition-state regions solely by training on hitherto popular benchmark datasets. Transition1x is a new challenging benchmark that will provide an important step towards developing next-generation ML force fields that also work far away from equilibrium configurations and reactive systems.
NeuralNEB -- Neural Networks can find Reaction Paths Fast
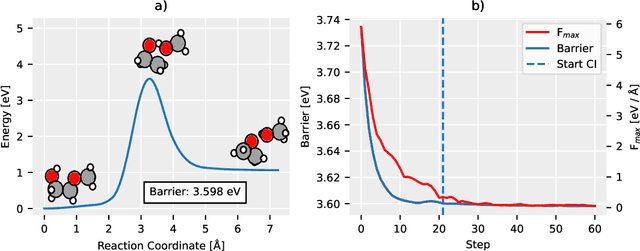
Jul 21, 2022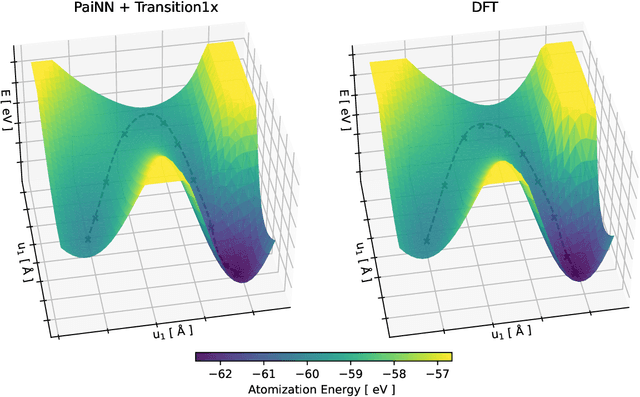
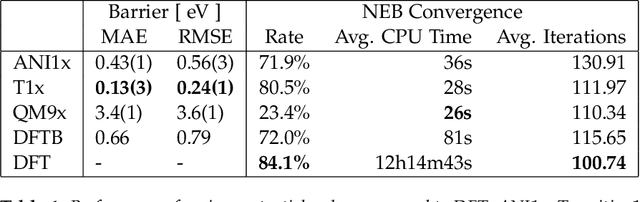
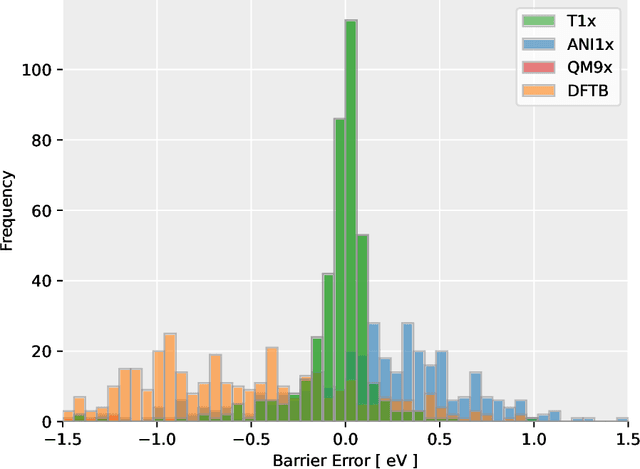
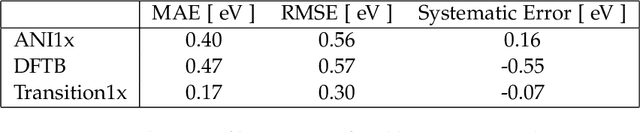
Quantum mechanical methods like Density Functional Theory (DFT) are used with great success alongside efficient search algorithms for studying kinetics of reactive systems. However, DFT is prohibitively expensive for large scale exploration. Machine Learning (ML) models have turned out to be excellent emulators of small molecule DFT calculations and could possibly replace DFT in such tasks. For kinetics, success relies primarily on the models capability to accurately predict the Potential Energy Surface (PES) around transition-states and Minimal Energy Paths (MEPs). Previously this has not been possible due to scarcity of relevant data in the literature. In this paper we train state of the art equivariant Graph Neural Network (GNN)-based models on around 10.000 elementary reactions from the Transition1x dataset. We apply the models as potentials for the Nudged Elastic Band (NEB) algorithm and achieve a Mean Average Error (MAE) of 0.13+/-0.03 eV on barrier energies on unseen reactions. We compare the results against equivalent models trained on QM9 and ANI1x. We also compare with and outperform Density Functional based Tight Binding (DFTB) on both accuracy and computational resource. The implication is that ML models, given relevant data, are now at a level where they can be applied for downstream tasks in quantum chemistry transcending prediction of simple molecular features.
Calibrated Uncertainty for Molecular Property Prediction using Ensembles of Message Passing Neural Networks
Jul 13, 2021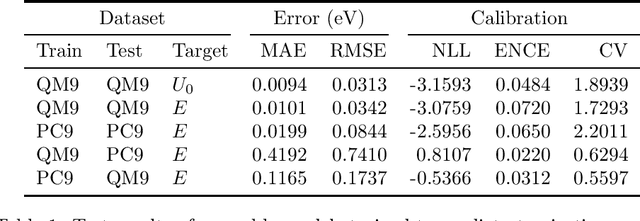

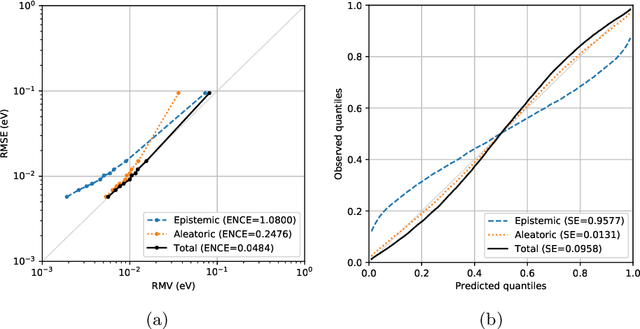
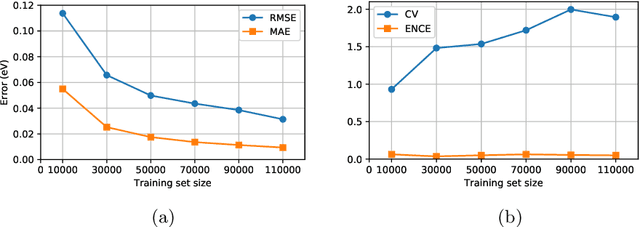
Data-driven methods based on machine learning have the potential to accelerate analysis of atomic structures. However, machine learning models can produce overconfident predictions and it is therefore crucial to detect and handle uncertainty carefully. Here, we extend a message passing neural network designed specifically for predicting properties of molecules and materials with a calibrated probabilistic predictive distribution. The method presented in this paper differs from the previous work by considering both aleatoric and epistemic uncertainty in a unified framework, and by re-calibrating the predictive distribution on unseen data. Through computer experiments, we show that our approach results in accurate models for predicting molecular formation energies with calibrated uncertainty in and out of the training data distribution on two public molecular benchmark datasets, QM9 and PC9. The proposed method provides a general framework for training and evaluating neural network ensemble models that are able to produce accurate predictions of properties of molecules with calibrated uncertainty.