 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Knowledge to Action: Outcomes of the 2025 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry
May 04, 2026Large language models (LLMs) are rapidly changing how researchers in materials science and chemistry discover, organize, and act on scientific knowledge. This paper analyzes a broad set of community-developed LLM applications in an effort to identify emerging patterns in how these systems can be used across the scientific research lifecycle. We organize the projects into two complementary categories: Knowledge Infrastructure, systems that structure, retrieve, synthesize, and validate scientific information; and Action Systems, systems that execute, coordinate, or automate scientific work across computational and experimental environments. The submissions reveal a shift from single-purpose LLM tools toward integrated, multi-agent workflows that combine retrieval, reasoning, tool use, and domain-specific validation. Prominent themes include retrieval-augmented generation as grounding infrastructure, persistent structured knowledge representations, multimodal and multilingual scientific inputs, and early progress toward laboratory-integrated closed-loop systems. Together, these results suggest that LLMs are evolving from general-purpose assistants into composable infrastructure for scientific reasoning and action. This work provides a community snapshot of that transition and a practical taxonomy for understanding emerging LLM-enabled workflows in materials science and chemistry.
Symmetry-Constrained Generation of Diverse Low-Bandgap Molecules with Monte Carlo Tree Search
Oct 11, 2024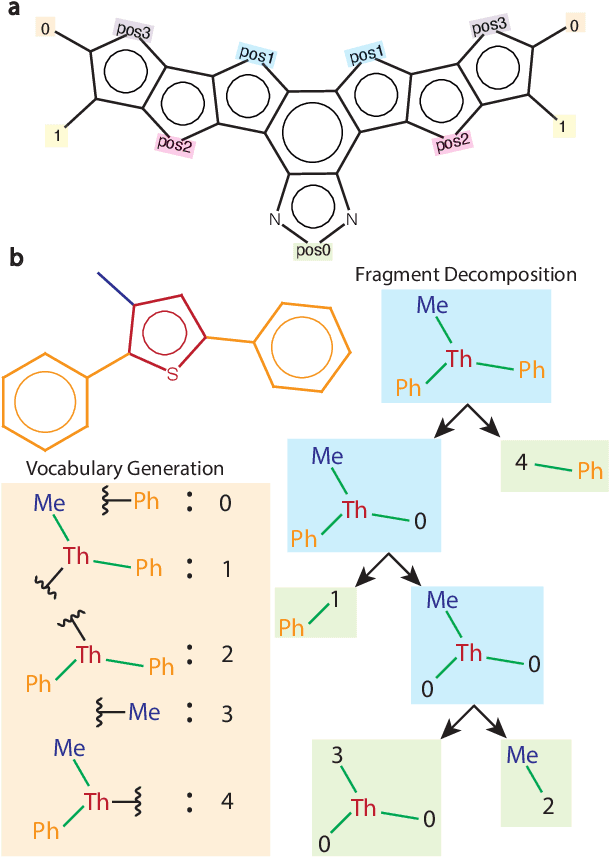
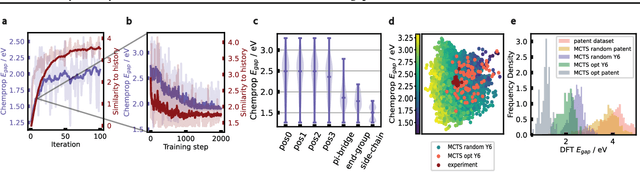
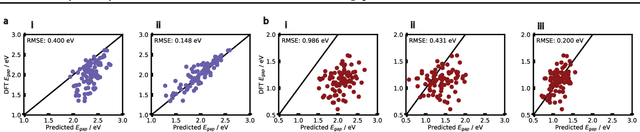
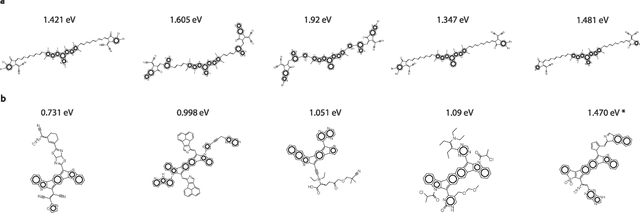
Organic optoelectronic materials are a promising avenue for next-generation electronic devices due to their solution processability, mechanical flexibility, and tunable electronic properties. In particular, near-infrared (NIR) sensitive molecules have unique applications in night-vision equipment and biomedical imaging. Molecular engineering has played a crucial role in developing non-fullerene acceptors (NFAs) such as the Y-series molecules, which have significantly improved the power conversion efficiency (PCE) of solar cells and enhanced spectral coverage in the NIR region. However, systematically designing molecules with targeted optoelectronic properties while ensuring synthetic accessibility remains a challenge. To address this, we leverage structural priors from domain-focused, patent-mined datasets of organic electronic molecules using a symmetry-aware fragment decomposition algorithm and a fragment-constrained Monte Carlo Tree Search (MCTS) generator. Our approach generates candidates that retain symmetry constraints from the patent dataset, while also exhibiting red-shifted absorption, as validated by TD-DFT calculations.
Efficient Generation of Molecular Clusters with Dual-Scale Equivariant Flow Matching
Oct 10, 2024Amorphous molecular solids offer a promising alternative to inorganic semiconductors, owing to their mechanical flexibility and solution processability. The packing structure of these materials plays a crucial role in determining their electronic and transport properties, which are key to enhancing the efficiency of devices like organic solar cells (OSCs). However, obtaining these optoelectronic properties computationally requires molecular dynamics (MD) simulations to generate a conformational ensemble, a process that can be computationally expensive due to the large system sizes involved. Recent advances have focused on using generative models, particularly flow-based models as Boltzmann generators, to improve the efficiency of MD sampling. In this work, we developed a dual-scale flow matching method that separates training and inference into coarse-grained and all-atom stages and enhances both the accuracy and efficiency of standard flow matching samplers. We demonstrate the effectiveness of this method on a dataset of Y6 molecular clusters obtained through MD simulations, and we benchmark its efficiency and accuracy against single-scale flow matching methods.
Automated patent extraction powers generative modeling in focused chemical spaces
Mar 14, 2023

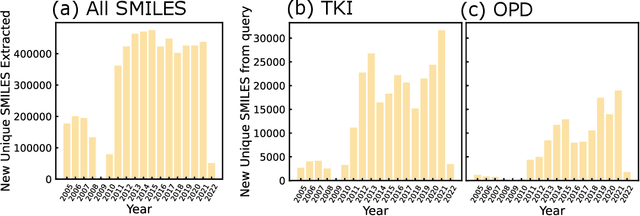

Deep generative models have emerged as an exciting avenue for inverse molecular design, with progress coming from the interplay between training algorithms and molecular representations. One of the key challenges in their applicability to materials science and chemistry has been the lack of access to sizeable training datasets with property labels. Published patents contain the first disclosure of new materials prior to their publication in journals, and are a vast source of scientific knowledge that has remained relatively untapped in the field of data-driven molecular design. Because patents are filed seeking to protect specific uses, molecules in patents can be considered to be weakly labeled into application classes. Furthermore, patents published by the US Patent and Trademark Office (USPTO) are downloadable and have machine-readable text and molecular structures. In this work, we train domain-specific generative models using patent data sources by developing an automated pipeline to go from USPTO patent digital files to the generation of novel candidates with minimal human intervention. We test the approach on two in-class extracted datasets, one in organic electronics and another in tyrosine kinase inhibitors. We then evaluate the ability of generative models trained on these in-class datasets on two categories of tasks (distribution learning and property optimization), identify strengths and limitations, and suggest possible explanations and remedies that could be used to overcome these in practice.
COVIDScholar: An automated COVID-19 research aggregation and analysis platform
Dec 07, 2020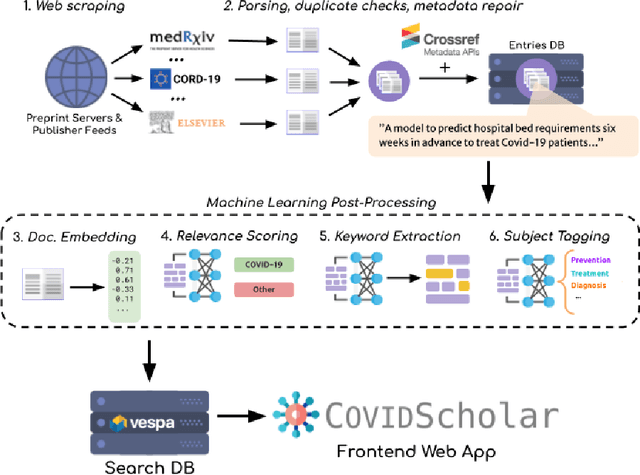
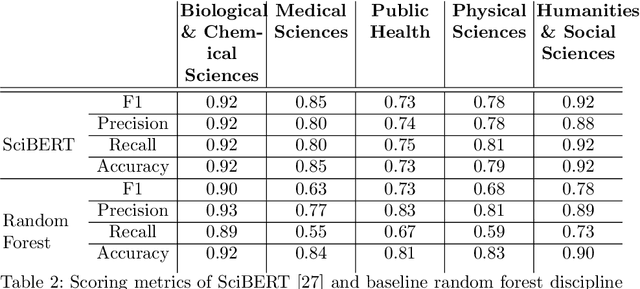
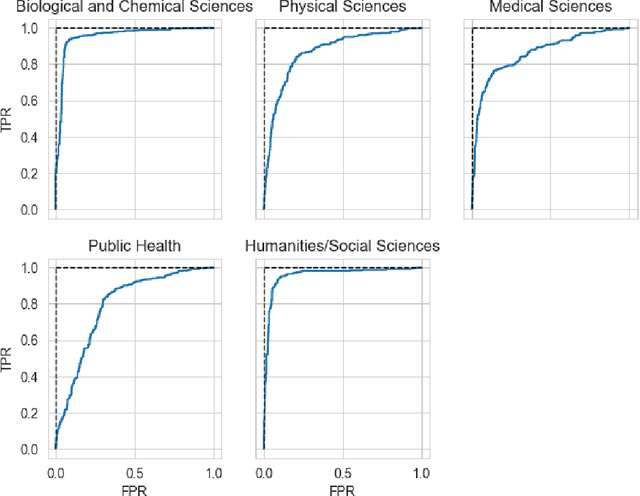
The ongoing COVID-19 pandemic has had far-reaching effects throughout society, and science is no exception. The scale, speed, and breadth of the scientific community's COVID-19 response has lead to the emergence of new research literature on a remarkable scale -- as of October 2020, over 81,000 COVID-19 related scientific papers have been released, at a rate of over 250 per day. This has created a challenge to traditional methods of engagement with the research literature; the volume of new research is far beyond the ability of any human to read, and the urgency of response has lead to an increasingly prominent role for pre-print servers and a diffusion of relevant research across sources. These factors have created a need for new tools to change the way scientific literature is disseminated. COVIDScholar is a knowledge portal designed with the unique needs of the COVID-19 research community in mind, utilizing NLP to aid researchers in synthesizing the information spread across thousands of emergent research articles, patents, and clinical trials into actionable insights and new knowledge. The search interface for this corpus, https://covidscholar.org, now serves over 2000 unique users weekly. We present also an analysis of trends in COVID-19 research over the course of 2020.
Inverse Design of Potential Singlet Fission Molecules using a Transfer Learning Based Approach
Mar 17, 2020
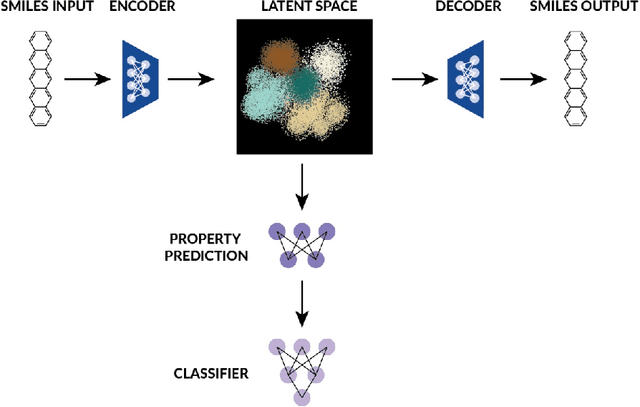
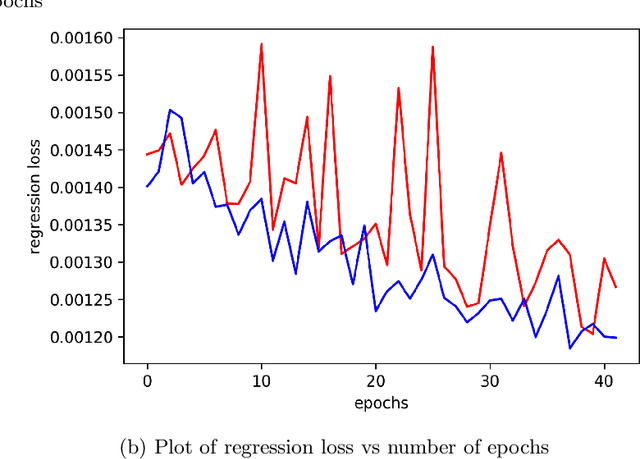
Singlet fission has emerged as one of the most exciting phenomena known to improve the efficiencies of different types of solar cells and has found uses in diverse optoelectronic applications. The range of available singlet fission molecules is, however, limited as to undergo singlet fission, molecules have to satisfy certain energy conditions. Recent advances in material search using inverse design has enabled the prediction of materials for a wide range of applications and has emerged as one of the most efficient methods in the discovery of suitable materials. It is particularly helpful in manipulating large datasets, uncovering hidden information from the molecular dataset and generating new structures. However, we seldom encounter large datasets in structure prediction problems in material science. In our work, we put forward inverse design of possible singlet fission molecules using a transfer learning based approach where we make use of a much larger ChEMBL dataset of structurally similar molecules to transfer the learned characteristics to the singlet fission dataset.