 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning Ordering in Crystalline Materials with Symmetry-Aware Graph Neural Networks
Sep 20, 2024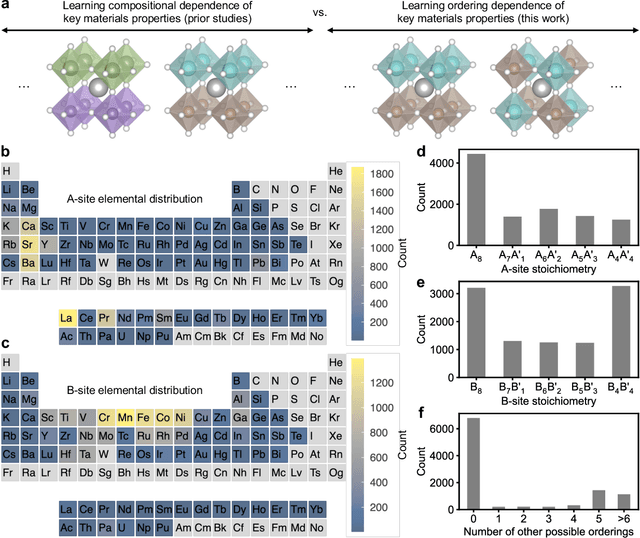
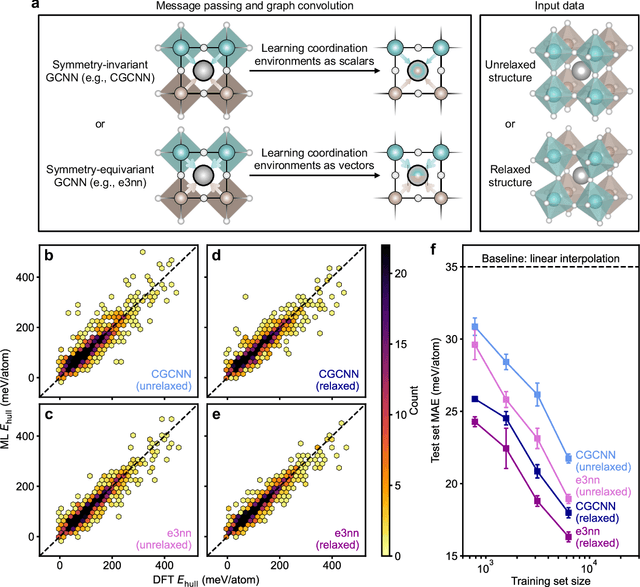
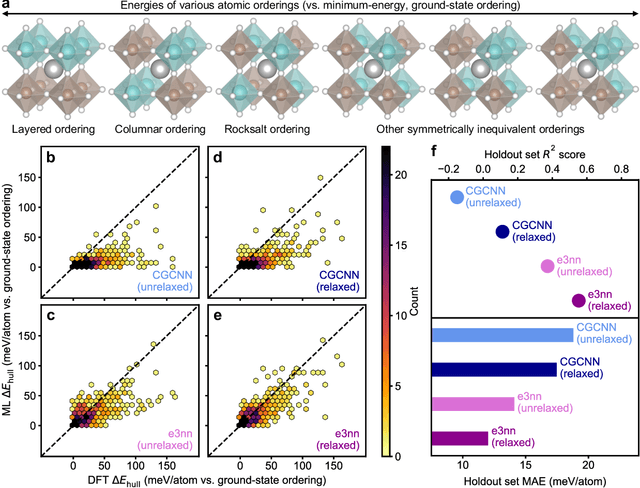
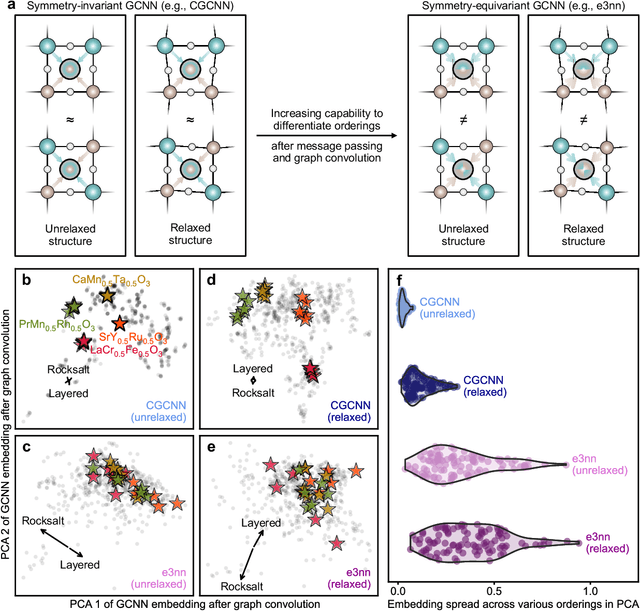
Graph convolutional neural networks (GCNNs) have become a machine learning workhorse for screening the chemical space of crystalline materials in fields such as catalysis and energy storage, by predicting properties from structures. Multicomponent materials, however, present a unique challenge since they can exhibit chemical (dis)order, where a given lattice structure can encompass a variety of elemental arrangements ranging from highly ordered structures to fully disordered solid solutions. Critically, properties like stability, strength, and catalytic performance depend not only on structures but also on orderings. To enable rigorous materials design, it is thus critical to ensure GCNNs are capable of distinguishing among atomic orderings. However, the ordering-aware capability of GCNNs has been poorly understood. Here, we benchmark various neural network architectures for capturing the ordering-dependent energetics of multicomponent materials in a custom-made dataset generated with high-throughput atomistic simulations. Conventional symmetry-invariant GCNNs were found unable to discern the structural difference between the diverse symmetrically inequivalent atomic orderings of the same material, while symmetry-equivariant model architectures could inherently preserve and differentiate the distinct crystallographic symmetries of various orderings.