 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAVIDa-hIL6: A Large-Scale VHH Dataset Produced from an Immunized Alpaca for Predicting Antigen-Antibody Interactions
Jun 06, 2023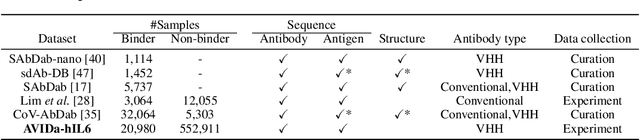
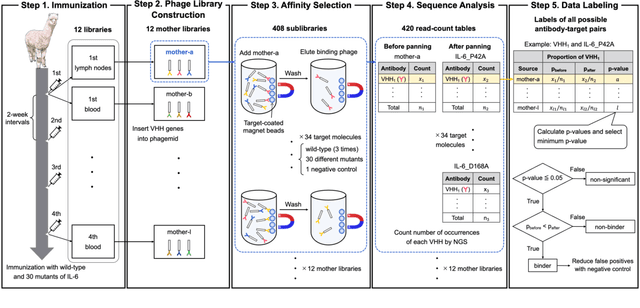
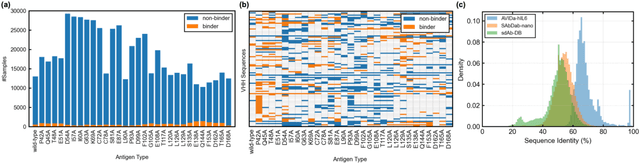

Antibodies have become an important class of therapeutic agents to treat human diseases. To accelerate therapeutic antibody discovery, computational methods, especially machine learning, have attracted considerable interest for predicting specific interactions between antibody candidates and target antigens such as viruses and bacteria. However, the publicly available datasets in existing works have notable limitations, such as small sizes and the lack of non-binding samples and exact amino acid sequences. To overcome these limitations, we have developed AVIDa-hIL6, a large-scale dataset for predicting antigen-antibody interactions in the variable domain of heavy chain of heavy chain antibodies (VHHs), produced from an alpaca immunized with the human interleukin-6 (IL-6) protein, as antigens. By leveraging the simple structure of VHHs, which facilitates identification of full-length amino acid sequences by DNA sequencing technology, AVIDa-hIL6 contains 573,891 antigen-VHH pairs with amino acid sequences. All the antigen-VHH pairs have reliable labels for binding or non-binding, as generated by a novel labeling method. Furthermore, via introduction of artificial mutations, AVIDa-hIL6 contains 30 different mutants in addition to wild-type IL-6 protein. This characteristic provides opportunities to develop machine learning models for predicting changes in antibody binding by antigen mutations. We report experimental benchmark results on AVIDa-hIL6 by using neural network-based baseline models. The results indicate that the existing models have potential, but further research is needed to generalize them to predict effective antibodies against unknown mutants. The dataset is available at https://avida-hil6.cognanous.com.
Meaningful machine learning models and machine-learned pharmacophores from fragment screening campaigns
Mar 25, 2022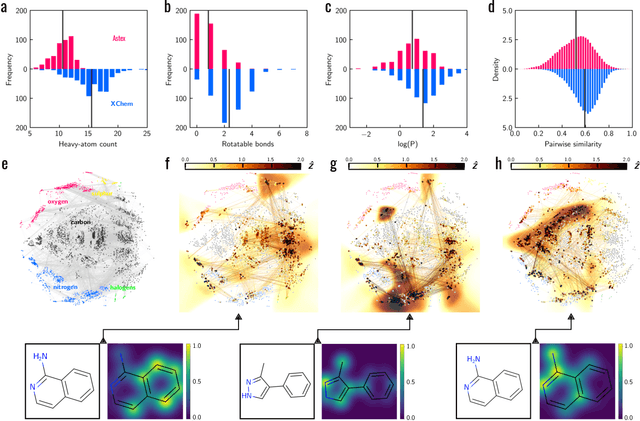
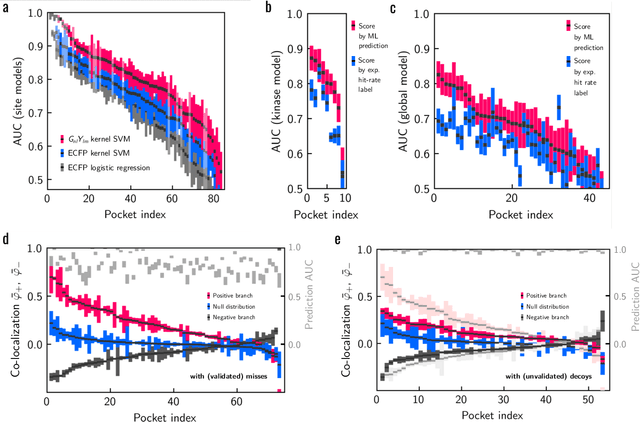
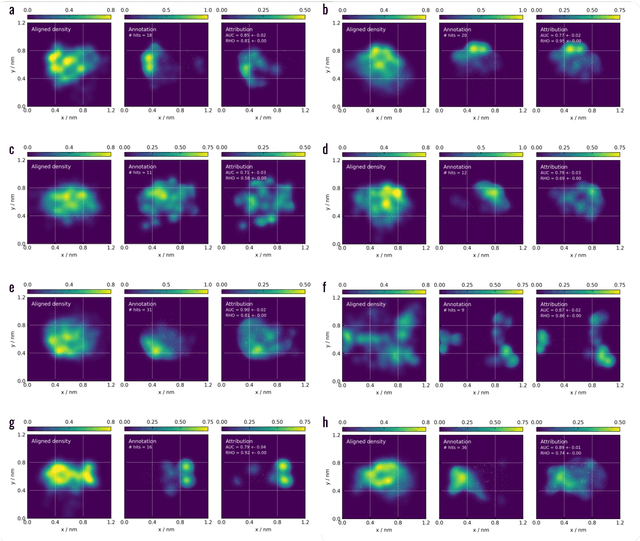
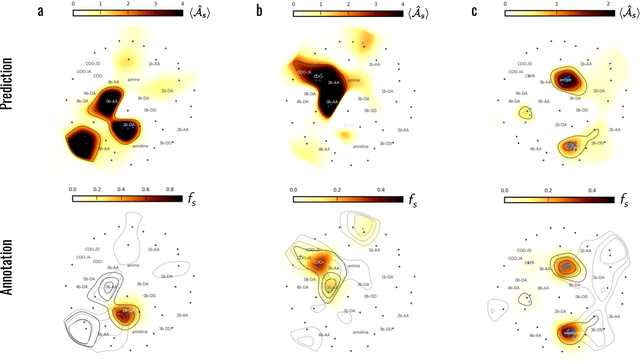
Machine learning (ML) is widely used in drug discovery to train models that predict protein-ligand binding. These models are of great value to medicinal chemists, in particular if they provide case-specific insight into the physical interactions that drive the binding process. In this study we derive ML models from over 50 fragment-screening campaigns to introduce two important elements that we believe are absent in most -- if not all -- ML studies of this type reported to date: First, alongside the observed hits we use to train our models, we incorporate true misses and show that these experimentally validated negative data are of significant importance to the quality of the derived models. Second, we provide a physically interpretable and verifiable representation of what the ML model considers important for successful binding. This representation is derived from a straightforward attribution procedure that explains the prediction in terms of the (inter-)action of chemical environments. Critically, we validate the attribution outcome on a large scale against prior annotations made independently by expert molecular modellers. We find good agreement between the key molecular substructures proposed by the ML model and those assigned manually, even when the model's performance in discriminating hits from misses is far from perfect. By projecting the attribution onto predefined interaction prototypes (pharmacophores), we show that ML allows us to formulate simple rules for what drives fragment binding against a target automatically from screening data.
Rethinking Attention with Performers
Sep 30, 2020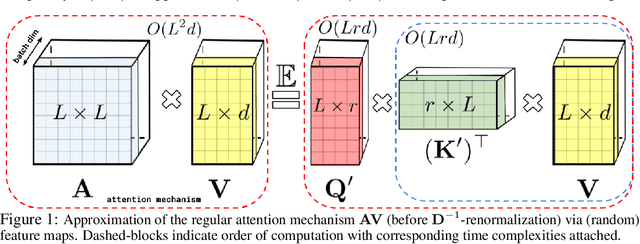
We introduce Performers, Transformer architectures which can estimate regular (softmax) full-rank-attention Transformers with provable accuracy, but using only linear (as opposed to quadratic) space and time complexity, without relying on any priors such as sparsity or low-rankness. To approximate softmax attention-kernels, Performers use a novel Fast Attention Via positive Orthogonal Random features approach (FAVOR+), which may be of independent interest for scalable kernel methods. FAVOR+ can be also used to efficiently model kernelizable attention mechanisms beyond softmax. This representational power is crucial to accurately compare softmax with other kernels for the first time on large-scale tasks, beyond the reach of regular Transformers, and investigate optimal attention-kernels. Performers are linear architectures fully compatible with regular Transformers and with strong theoretical guarantees: unbiased or nearly-unbiased estimation of the attention matrix, uniform convergence and low estimation variance. We tested Performers on a rich set of tasks stretching from pixel-prediction through text models to protein sequence modeling. We demonstrate competitive results with other examined efficient sparse and dense attention methods, showcasing effectiveness of the novel attention-learning paradigm leveraged by Performers.
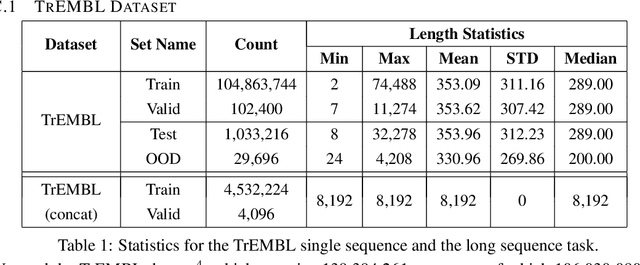
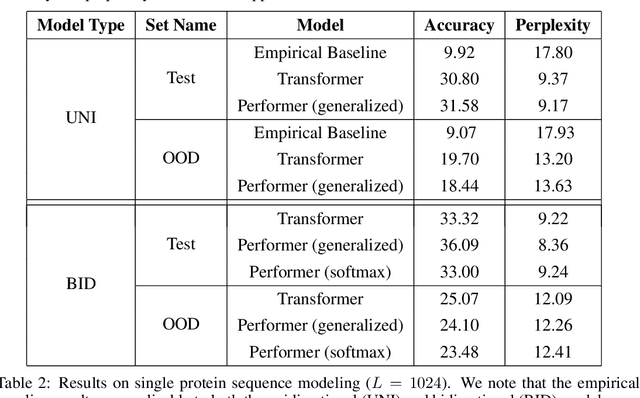
Masked Language Modeling for Proteins via Linearly Scalable Long-Context Transformers
Jun 05, 2020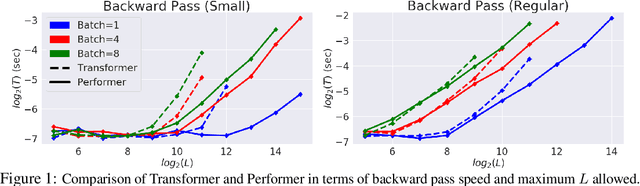
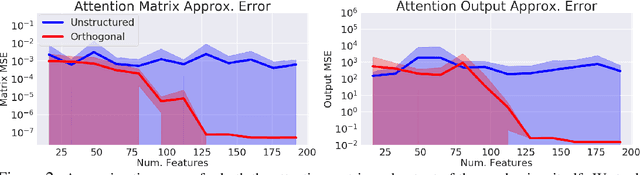
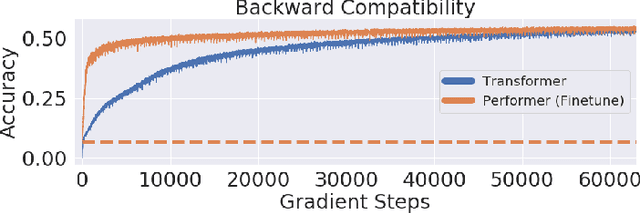

Transformer models have achieved state-of-the-art results across a diverse range of domains. However, concern over the cost of training the attention mechanism to learn complex dependencies between distant inputs continues to grow. In response, solutions that exploit the structure and sparsity of the learned attention matrix have blossomed. However, real-world applications that involve long sequences, such as biological sequence analysis, may fall short of meeting these assumptions, precluding exploration of these models. To address this challenge, we present a new Transformer architecture, Performer, based on Fast Attention Via Orthogonal Random features (FAVOR). Our mechanism scales linearly rather than quadratically in the number of tokens in the sequence, is characterized by sub-quadratic space complexity and does not incorporate any sparsity pattern priors. Furthermore, it provides strong theoretical guarantees: unbiased estimation of the attention matrix and uniform convergence. It is also backwards-compatible with pre-trained regular Transformers. We demonstrate its effectiveness on the challenging task of protein sequence modeling and provide detailed theoretical analysis.
Population-Based Black-Box Optimization for Biological Sequence Design
Jun 05, 2020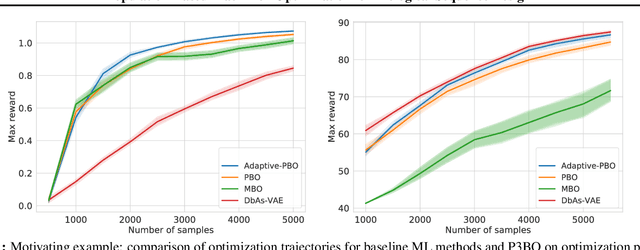
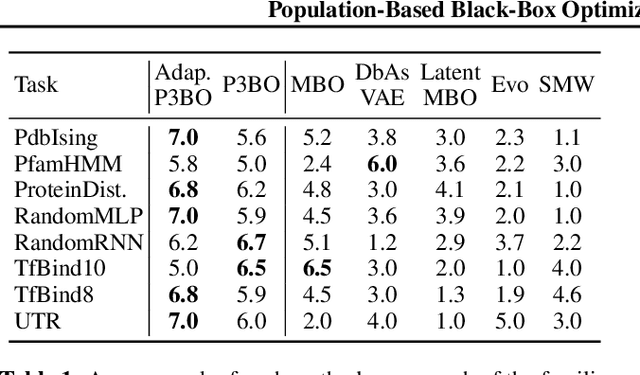
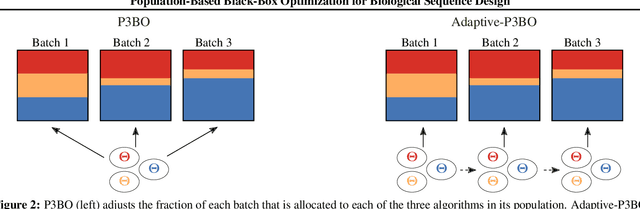
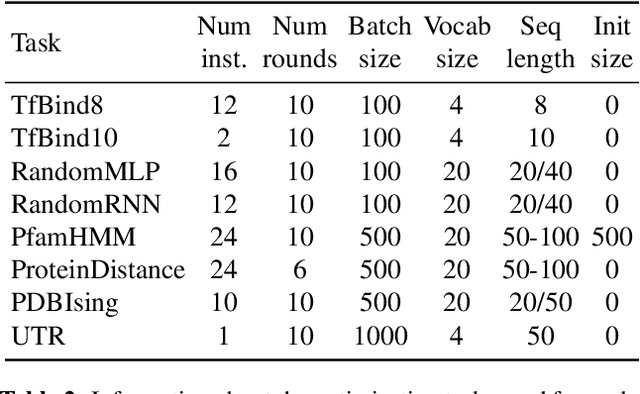
The use of black-box optimization for the design of new biological sequences is an emerging research area with potentially revolutionary impact. The cost and latency of wet-lab experiments requires methods that find good sequences in few experimental rounds of large batches of sequences--a setting that off-the-shelf black-box optimization methods are ill-equipped to handle. We find that the performance of existing methods varies drastically across optimization tasks, posing a significant obstacle to real-world applications. To improve robustness, we propose Population-Based Black-Box Optimization (P3BO), which generates batches of sequences by sampling from an ensemble of methods. The number of sequences sampled from any method is proportional to the quality of sequences it previously proposed, allowing P3BO to combine the strengths of individual methods while hedging against their innate brittleness. Adapting the hyper-parameters of each of the methods online using evolutionary optimization further improves performance. Through extensive experiments on in-silico optimization tasks, we show that P3BO outperforms any single method in its population, proposing higher quality sequences as well as more diverse batches. As such, P3BO and Adaptive-P3BO are a crucial step towards deploying ML to real-world sequence design.
Noisy, sparse, nonlinear: Navigating the Bermuda Triangle of physical inference with deep filtering
Nov 19, 2019
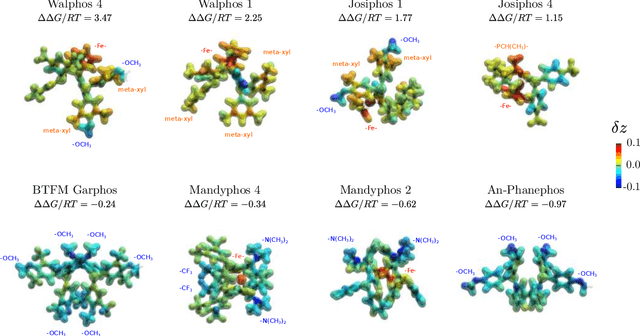
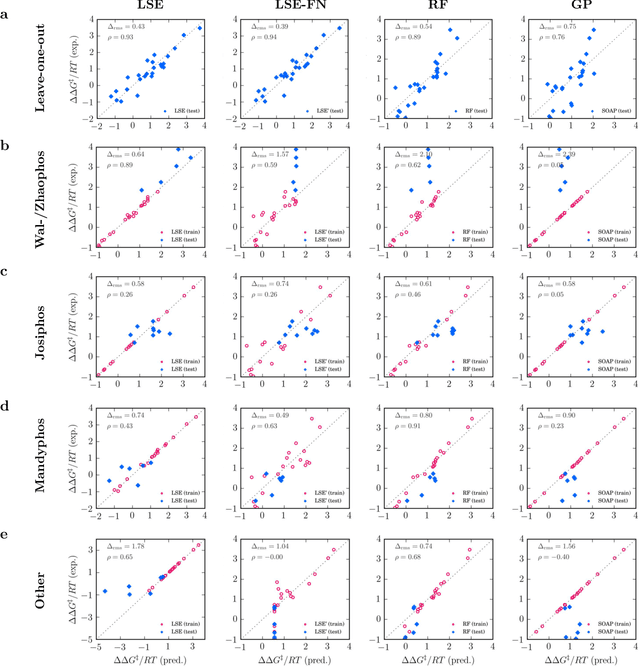
Capturing the microscopic interactions that determine molecular reactivity poses a challenge across the physical sciences. Even a basic understanding of the underlying reaction mechanisms can substantially accelerate materials and compound design, including the development of new catalysts or drugs. Given the difficulties routinely faced by both experimental and theoretical investigations that aim to improve our mechanistic understanding of a reaction, recent advances have focused on data-driven routes to derive structure-property relationships directly from high-throughput screens. However, even these high-quality, high-volume data are noisy, sparse and biased -- placing them in a regime where machine-learning is extremely challenging. Here we show that a statistical approach based on deep filtering of nonlinear feature networks results in physicochemical models that are more robust, transparent and generalize better than standard machine-learning architectures. Using diligent descriptor design and data post-processing, we exemplify the approach using both literature and fresh data on asymmetric catalytic hydrogenation, Palladium-catalyzed cross-coupling reactions, and drug-drug synergy. We illustrate how the sparse models uncovered by the filtering help us formulate physicochemical reaction ``pharmacophores'', investigate experimental bias and derive strategies for mechanism detection and classification.
Using Attribution to Decode Dataset Bias in Neural Network Models for Chemistry
Nov 29, 2018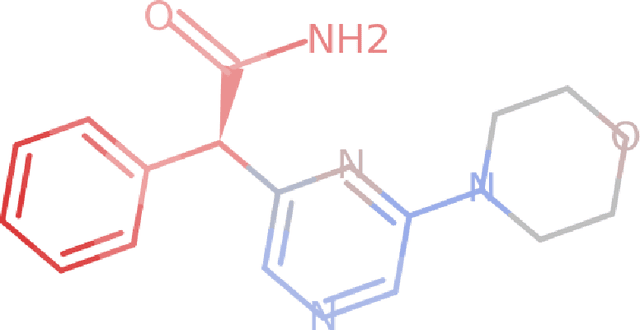
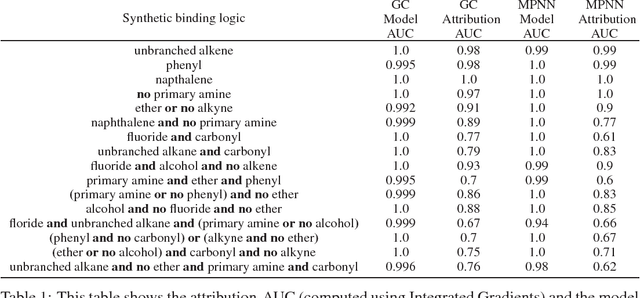
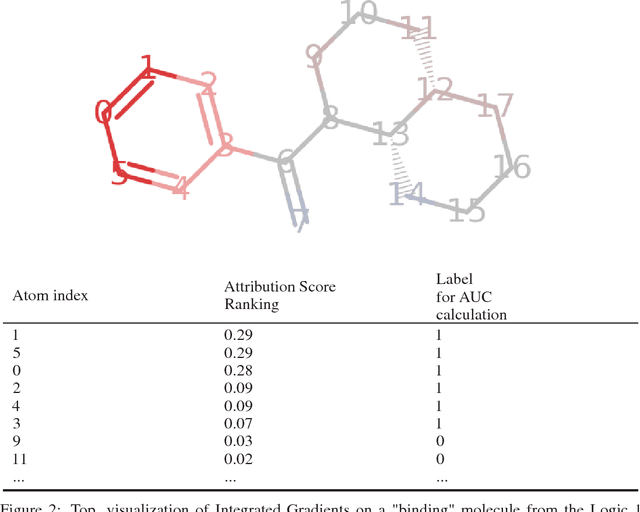
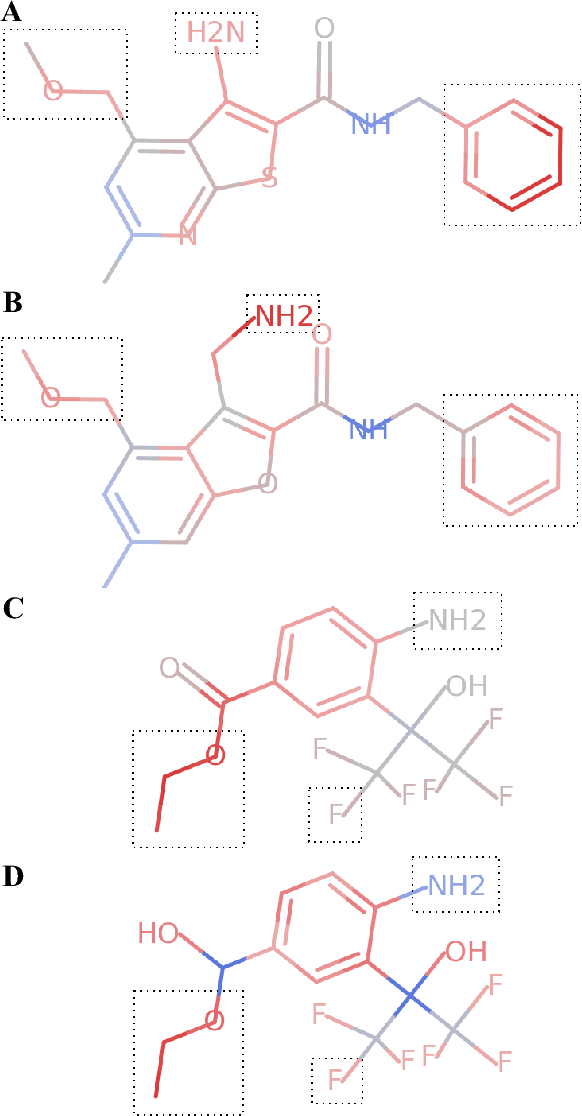
Deep neural networks have achieved state of the art accuracy at classifying molecules with respect to whether they bind to specific protein targets. A key breakthrough would occur if these models could reveal the fragment pharmacophores that are causally involved in binding. Extracting chemical details of binding from the networks could potentially lead to scientific discoveries about the mechanisms of drug actions. But doing so requires shining light into the black box that is the trained neural network model, a task that has proved difficult across many domains. Here we show how the binding mechanism learned by deep neural network models can be interrogated, using a recently described attribution method. We first work with carefully constructed synthetic datasets, in which the 'fragment logic' of binding is fully known. We find that networks that achieve perfect accuracy on held out test datasets still learn spurious correlations due to biases in the datasets, and we are able to exploit this non-robustness to construct adversarial examples that fool the model. The dataset bias makes these models unreliable for accurately revealing information about the mechanisms of protein-ligand binding. In light of our findings, we prescribe a test that checks for dataset bias given a hypothesis. If the test fails, it indicates that either the model must be simplified or regularized and/or that the training dataset requires augmentation.