 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEmbracing assay heterogeneity with neural processes for markedly improved bioactivity predictions
Aug 17, 2023



Predicting the bioactivity of a ligand is one of the hardest and most important challenges in computer-aided drug discovery. Despite years of data collection and curation efforts by research organizations worldwide, bioactivity data remains sparse and heterogeneous, thus hampering efforts to build predictive models that are accurate, transferable and robust. The intrinsic variability of the experimental data is further compounded by data aggregation practices that neglect heterogeneity to overcome sparsity. Here we discuss the limitations of these practices and present a hierarchical meta-learning framework that exploits the information synergy across disparate assays by successfully accounting for assay heterogeneity. We show that the model achieves a drastic improvement in affinity prediction across diverse protein targets and assay types compared to conventional baselines. It can quickly adapt to new target contexts using very few observations, thus enabling large-scale virtual screening in early-phase drug discovery.
3D pride without 2D prejudice: Bias-controlled multi-level generative models for structure-based ligand design
Apr 22, 2022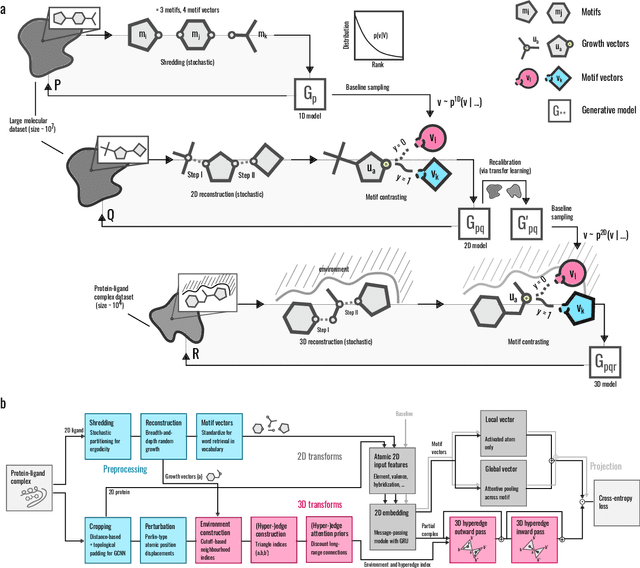
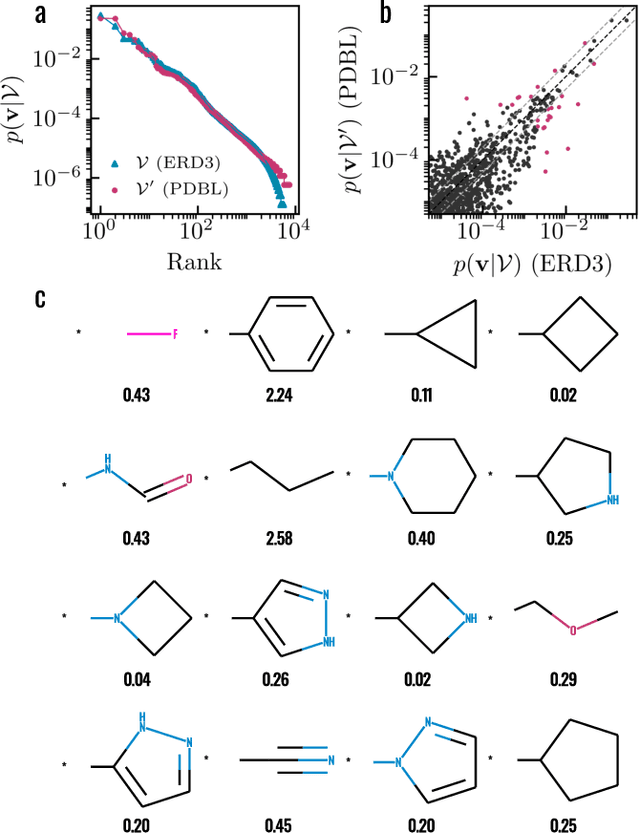
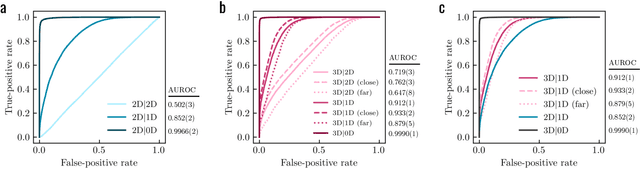
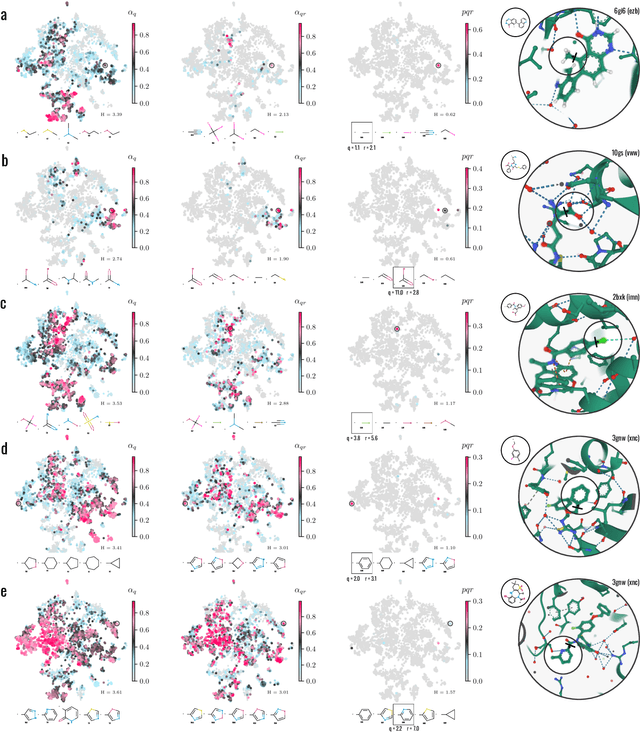
Generative models for structure-based molecular design hold significant promise for drug discovery, with the potential to speed up the hit-to-lead development cycle, while improving the quality of drug candidates and reducing costs. Data sparsity and bias are, however, two main roadblocks to the development of 3D-aware models. Here we propose a first-in-kind training protocol based on multi-level contrastive learning for improved bias control and data efficiency. The framework leverages the large data resources available for 2D generative modelling with datasets of ligand-protein complexes. The result are hierarchical generative models that are topologically unbiased, explainable and customizable. We show how, by deconvolving the generative posterior into chemical, topological and structural context factors, we not only avoid common pitfalls in the design and evaluation of generative models, but furthermore gain detailed insight into the generative process itself. This improved transparency significantly aids method development, besides allowing fine-grained control over novelty vs familiarity.
Meaningful machine learning models and machine-learned pharmacophores from fragment screening campaigns
Mar 25, 2022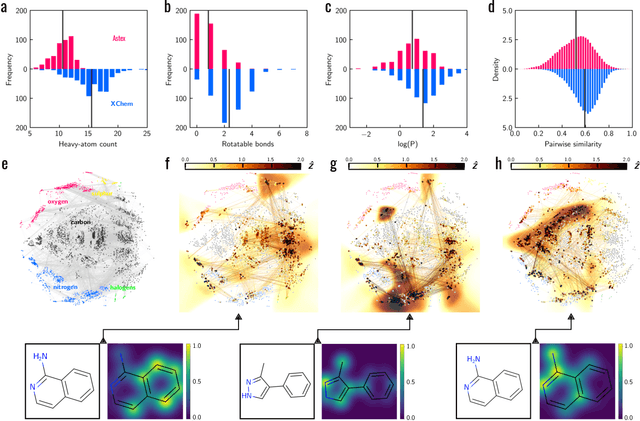
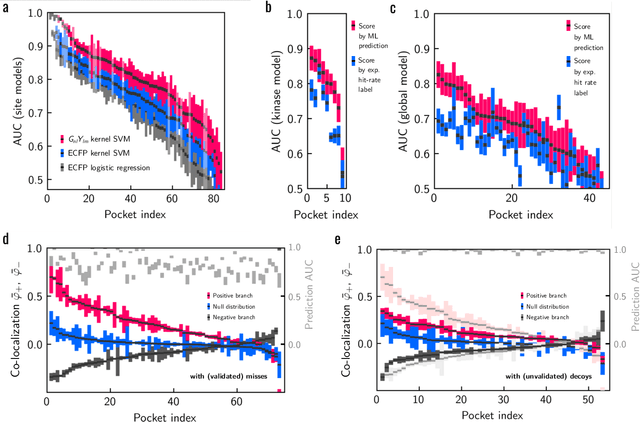
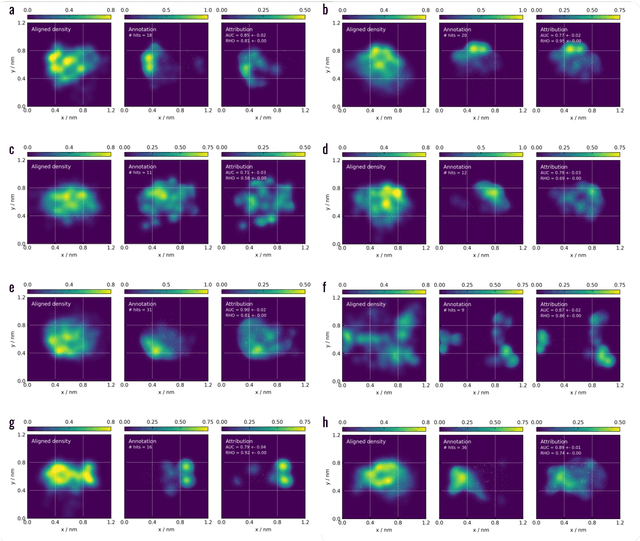
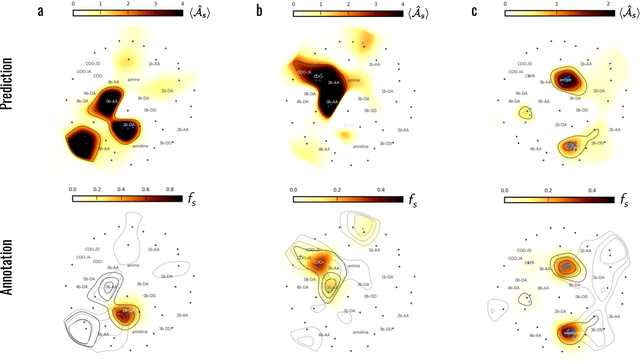
Machine learning (ML) is widely used in drug discovery to train models that predict protein-ligand binding. These models are of great value to medicinal chemists, in particular if they provide case-specific insight into the physical interactions that drive the binding process. In this study we derive ML models from over 50 fragment-screening campaigns to introduce two important elements that we believe are absent in most -- if not all -- ML studies of this type reported to date: First, alongside the observed hits we use to train our models, we incorporate true misses and show that these experimentally validated negative data are of significant importance to the quality of the derived models. Second, we provide a physically interpretable and verifiable representation of what the ML model considers important for successful binding. This representation is derived from a straightforward attribution procedure that explains the prediction in terms of the (inter-)action of chemical environments. Critically, we validate the attribution outcome on a large scale against prior annotations made independently by expert molecular modellers. We find good agreement between the key molecular substructures proposed by the ML model and those assigned manually, even when the model's performance in discriminating hits from misses is far from perfect. By projecting the attribution onto predefined interaction prototypes (pharmacophores), we show that ML allows us to formulate simple rules for what drives fragment binding against a target automatically from screening data.
BenchML: an extensible pipelining framework for benchmarking representations of materials and molecules at scale
Dec 04, 2021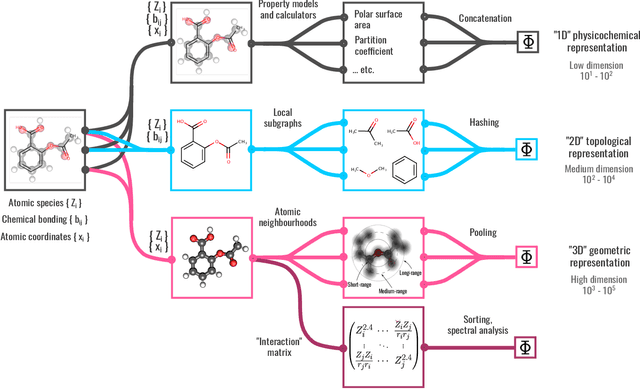
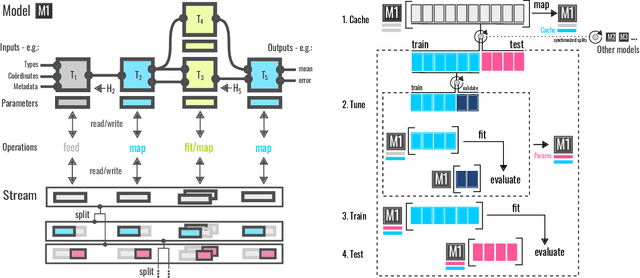
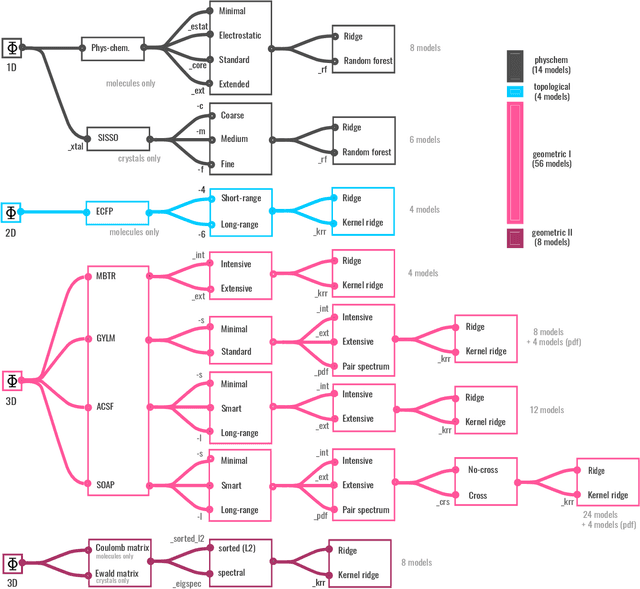
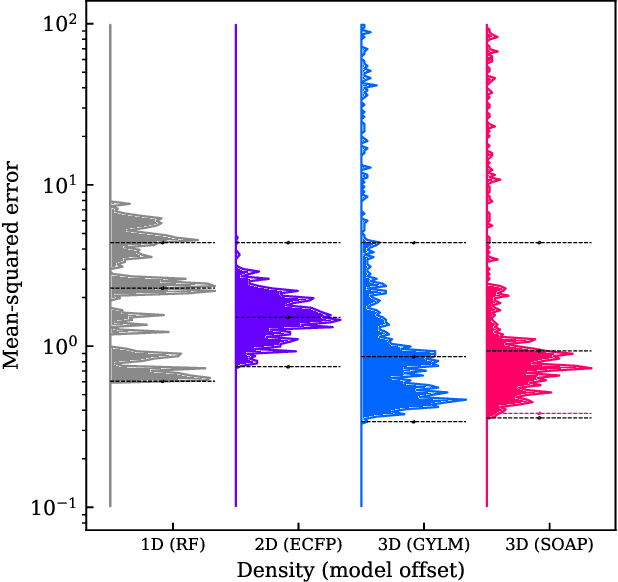
We introduce a machine-learning (ML) framework for high-throughput benchmarking of diverse representations of chemical systems against datasets of materials and molecules. The guiding principle underlying the benchmarking approach is to evaluate raw descriptor performance by limiting model complexity to simple regression schemes while enforcing best ML practices, allowing for unbiased hyperparameter optimization, and assessing learning progress through learning curves along series of synchronized train-test splits. The resulting models are intended as baselines that can inform future method development, next to indicating how easily a given dataset can be learnt. Through a comparative analysis of the training outcome across a diverse set of physicochemical, topological and geometric representations, we glean insight into the relative merits of these representations as well as their interrelatedness.
Investigating 3D Atomic Environments for Enhanced QSAR
Oct 24, 2020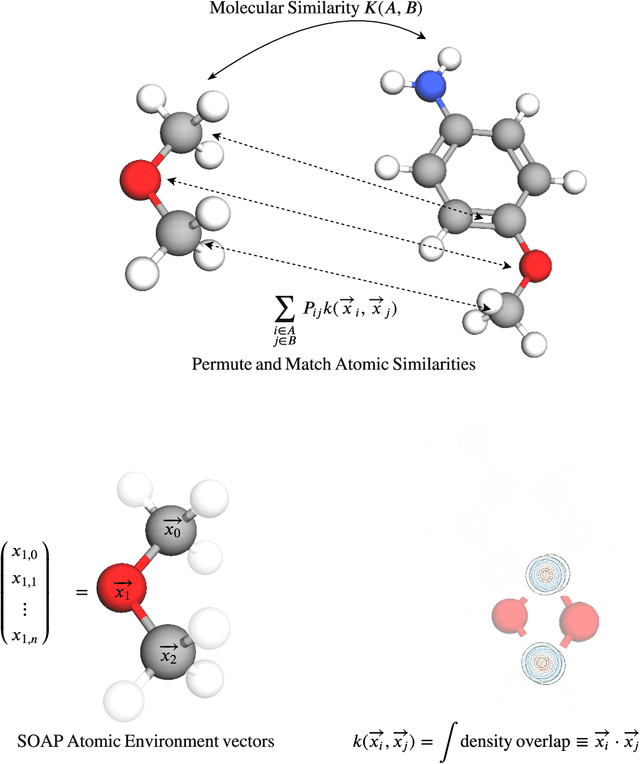
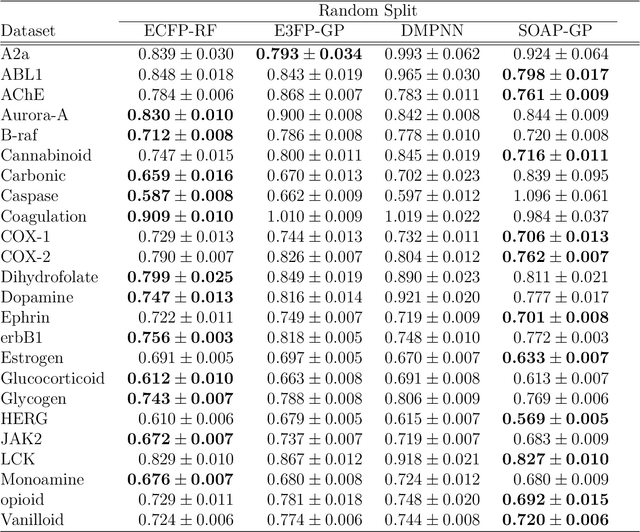
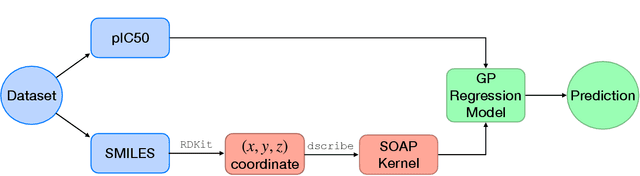
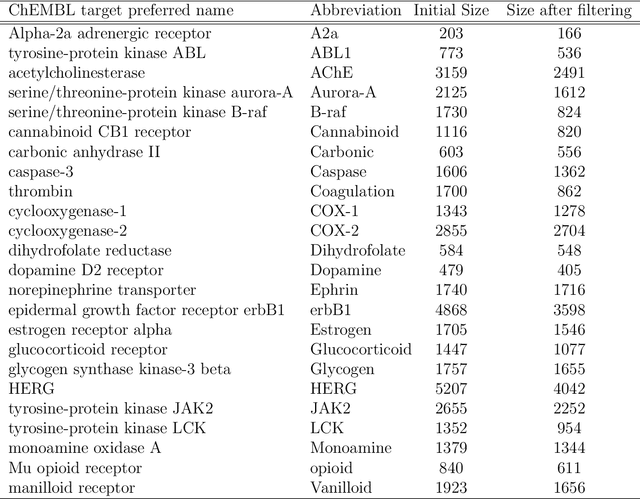
Predicting bioactivity and physical properties of molecules is a longstanding challenge in drug design. Most approaches use molecular descriptors based on a 2D representation of molecules as a graph of atoms and bonds, abstracting away the molecular shape. A difficulty in accounting for 3D shape is in designing molecular descriptors can precisely capture molecular shape while remaining invariant to rotations/translations. We describe a novel alignment-free 3D QSAR method using Smooth Overlap of Atomic Positions (SOAP), a well-established formalism developed for interpolating potential energy surfaces. We show that this approach rigorously describes local 3D atomic environments to compare molecular shapes in a principled manner. This method performs competitively with traditional fingerprint-based approaches as well as state-of-the-art graph neural networks on pIC$_{50}$ ligand-binding prediction in both random and scaffold split scenarios. We illustrate the utility of SOAP descriptors by showing that its inclusion in ensembling diverse representations statistically improves performance, demonstrating that incorporating 3D atomic environments could lead to enhanced QSAR for cheminformatics.
Noisy, sparse, nonlinear: Navigating the Bermuda Triangle of physical inference with deep filtering
Nov 19, 2019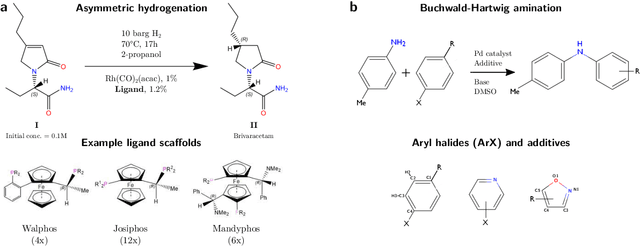
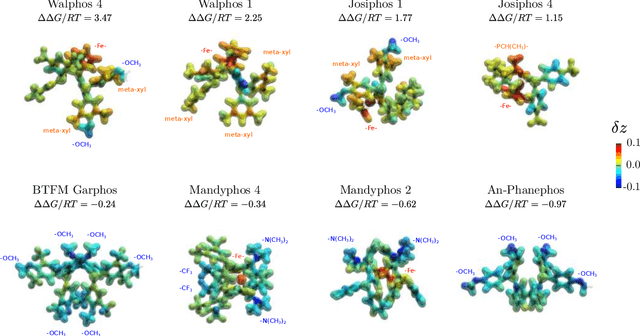
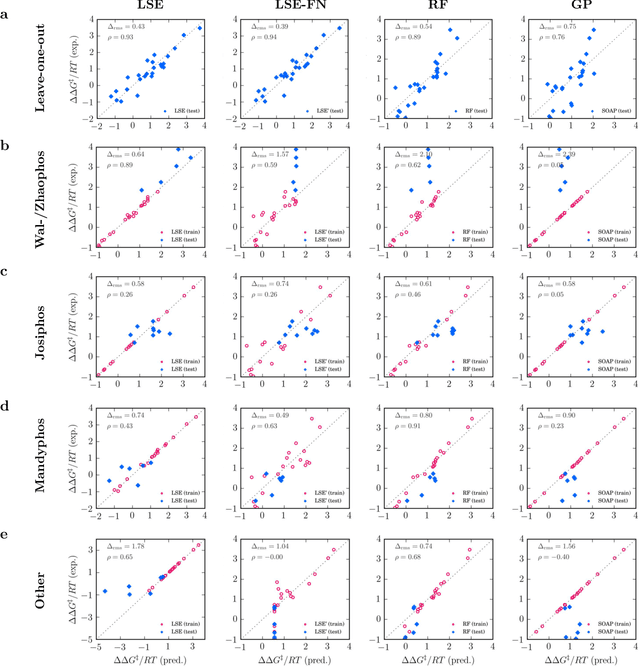
Capturing the microscopic interactions that determine molecular reactivity poses a challenge across the physical sciences. Even a basic understanding of the underlying reaction mechanisms can substantially accelerate materials and compound design, including the development of new catalysts or drugs. Given the difficulties routinely faced by both experimental and theoretical investigations that aim to improve our mechanistic understanding of a reaction, recent advances have focused on data-driven routes to derive structure-property relationships directly from high-throughput screens. However, even these high-quality, high-volume data are noisy, sparse and biased -- placing them in a regime where machine-learning is extremely challenging. Here we show that a statistical approach based on deep filtering of nonlinear feature networks results in physicochemical models that are more robust, transparent and generalize better than standard machine-learning architectures. Using diligent descriptor design and data post-processing, we exemplify the approach using both literature and fresh data on asymmetric catalytic hydrogenation, Palladium-catalyzed cross-coupling reactions, and drug-drug synergy. We illustrate how the sparse models uncovered by the filtering help us formulate physicochemical reaction ``pharmacophores'', investigate experimental bias and derive strategies for mechanism detection and classification.