 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEmbracing assay heterogeneity with neural processes for markedly improved bioactivity predictions
Aug 17, 2023



Predicting the bioactivity of a ligand is one of the hardest and most important challenges in computer-aided drug discovery. Despite years of data collection and curation efforts by research organizations worldwide, bioactivity data remains sparse and heterogeneous, thus hampering efforts to build predictive models that are accurate, transferable and robust. The intrinsic variability of the experimental data is further compounded by data aggregation practices that neglect heterogeneity to overcome sparsity. Here we discuss the limitations of these practices and present a hierarchical meta-learning framework that exploits the information synergy across disparate assays by successfully accounting for assay heterogeneity. We show that the model achieves a drastic improvement in affinity prediction across diverse protein targets and assay types compared to conventional baselines. It can quickly adapt to new target contexts using very few observations, thus enabling large-scale virtual screening in early-phase drug discovery.
3D pride without 2D prejudice: Bias-controlled multi-level generative models for structure-based ligand design
Apr 22, 2022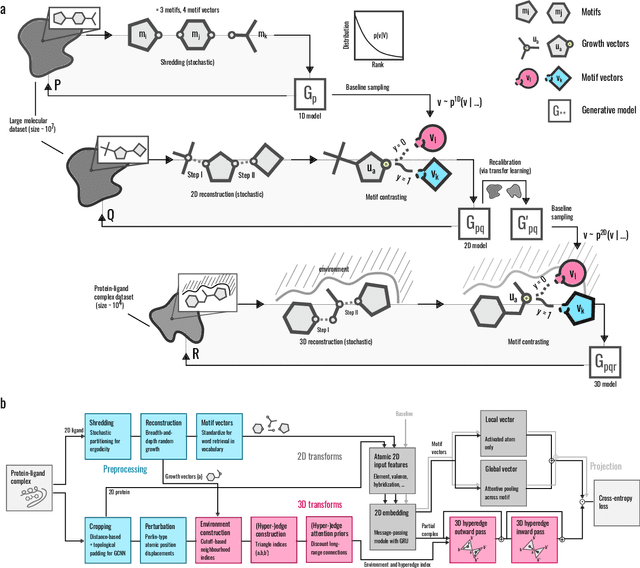
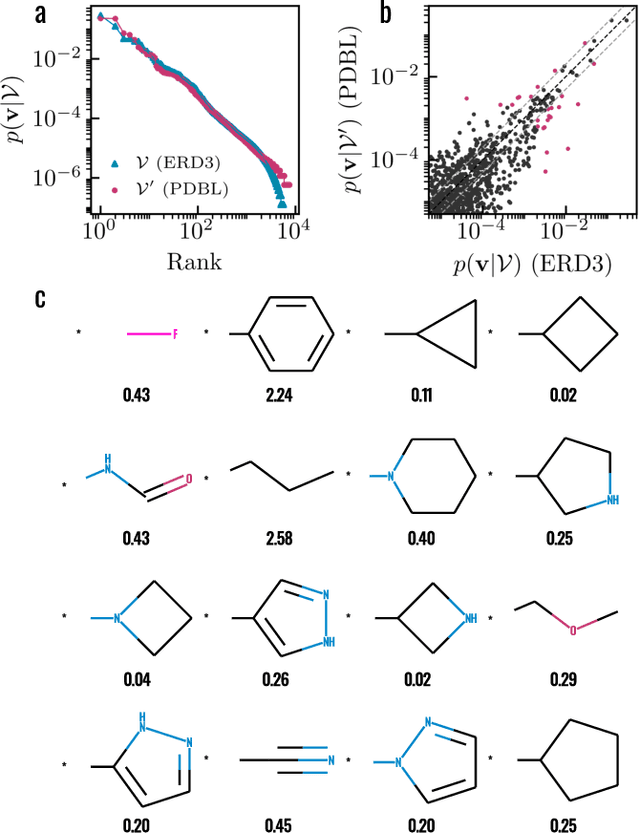
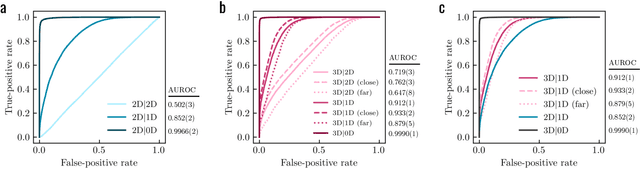
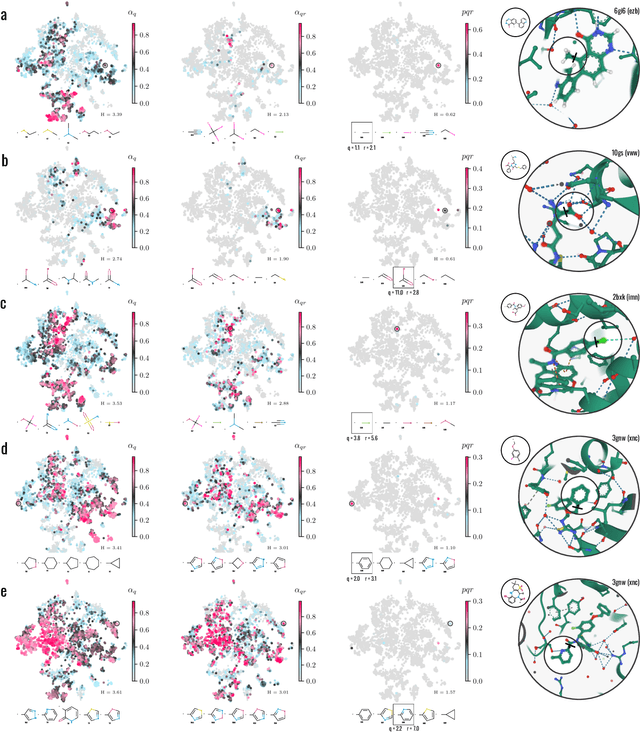
Generative models for structure-based molecular design hold significant promise for drug discovery, with the potential to speed up the hit-to-lead development cycle, while improving the quality of drug candidates and reducing costs. Data sparsity and bias are, however, two main roadblocks to the development of 3D-aware models. Here we propose a first-in-kind training protocol based on multi-level contrastive learning for improved bias control and data efficiency. The framework leverages the large data resources available for 2D generative modelling with datasets of ligand-protein complexes. The result are hierarchical generative models that are topologically unbiased, explainable and customizable. We show how, by deconvolving the generative posterior into chemical, topological and structural context factors, we not only avoid common pitfalls in the design and evaluation of generative models, but furthermore gain detailed insight into the generative process itself. This improved transparency significantly aids method development, besides allowing fine-grained control over novelty vs familiarity.
Meaningful machine learning models and machine-learned pharmacophores from fragment screening campaigns
Mar 25, 2022
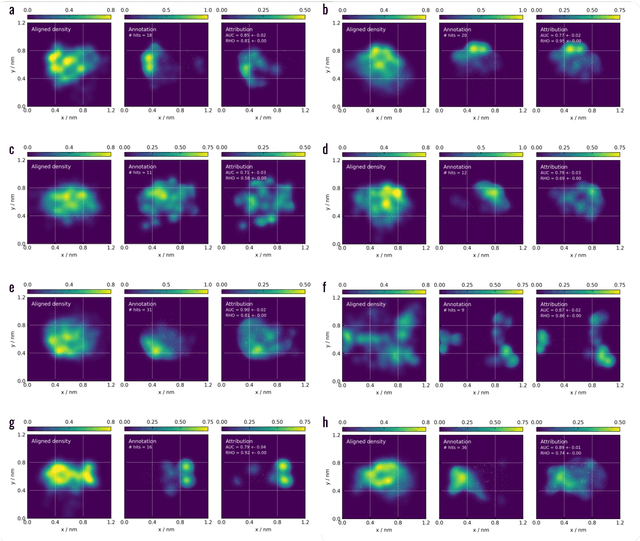
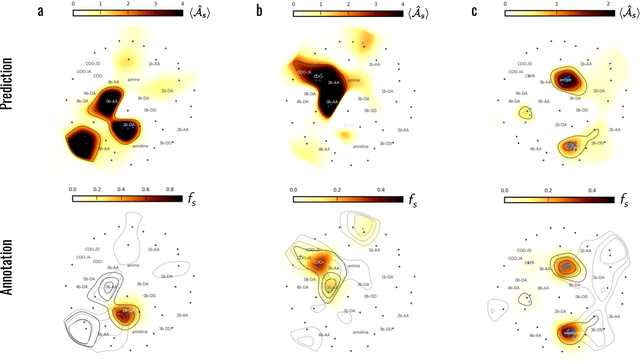
Machine learning (ML) is widely used in drug discovery to train models that predict protein-ligand binding. These models are of great value to medicinal chemists, in particular if they provide case-specific insight into the physical interactions that drive the binding process. In this study we derive ML models from over 50 fragment-screening campaigns to introduce two important elements that we believe are absent in most -- if not all -- ML studies of this type reported to date: First, alongside the observed hits we use to train our models, we incorporate true misses and show that these experimentally validated negative data are of significant importance to the quality of the derived models. Second, we provide a physically interpretable and verifiable representation of what the ML model considers important for successful binding. This representation is derived from a straightforward attribution procedure that explains the prediction in terms of the (inter-)action of chemical environments. Critically, we validate the attribution outcome on a large scale against prior annotations made independently by expert molecular modellers. We find good agreement between the key molecular substructures proposed by the ML model and those assigned manually, even when the model's performance in discriminating hits from misses is far from perfect. By projecting the attribution onto predefined interaction prototypes (pharmacophores), we show that ML allows us to formulate simple rules for what drives fragment binding against a target automatically from screening data.