 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDistilling latent electrostatics from foundation machine learning interatomic potentials
Jun 12, 2026Foundation machine learning interatomic potentials (MLIPs) have enabled atomistic simulations across broad regions of chemical and materials space, but many remain computationally expensive and lack explicit electrostatics, limiting their use for systems governed by long-range interactions and electrical response. Previously, we introduced Latent Ewald Summation (LES), which learns latent atomic charges and long-range electrostatics from density functional theory (DFT) energy and force labels alone. Here, we use LES to extract electrostatics that are latent in foundation models: energies and forces predicted by a teacher model are used to train a lightweight LES-augmented student MLIP, with optional fine-tuning on additional DFT data. The resulting models reduce computational cost while providing access to Born effective charge tensors, and infrared spectra. We benchmark student models distilled from a broad set of foundation MLIPs, including UMA, MACE, Orb, eSEN, GemNet-OC, PET, and EquiformerV2-based models, against experimental infrared spectra for liquid water, concentrated hydrochloric acid, and the anatase TiO2(101)-water interface. Across these systems, electrostatic response can be extracted from most foundation MLIPs. The benchmark further shows that the underlying DFT level and dataset used to train the teacher model play a larger role than architecture in determining electrostatic and spectroscopic accuracy. For the TiO2-water interface, fine-tuning with a modest amount of higher-level DFT data improves structural and infrared predictions. LES-based distillation therefore provides a practical route for converting foundation MLIPs into efficient, electrically responsive models, while also testing the physical fidelity encoded in foundation models.
Polarizable atomic multipoles for learning long-range electrostatics
May 07, 2026Long-range electrostatics and polarization remain central obstacles to extending machine learning interatomic potentials (MLIPs) to ionic, polar, and interfacial systems. Here, we introduce a semi-local framework for learning electrostatics from energies and forces using polarizable atomic multipoles. Local equivariant descriptors predict environment-dependent latent monopoles, dipoles, and quadrupoles, while residual non-local charge transfer and polarization are captured by non-self-consistent linear response in induced charges and dipoles. Across four diverse benchmarks and four short-range MLIP architectures, the multipole hierarchy and response terms systematically improve potential energy surface accuracy, with the largest gains in systems where long-range effects are essential. More importantly, the learned latent variables recover physically meaningful electrical responses: accurate Born effective charge tensors, emergent polarizabilities, infrared spectra in close agreement with experiments, and semi-quantitative Raman spectra for bulk water and hybrid MAPbI$_3$ perovskite. This systematically improvable, physically transparent framework enables MLIPs trained on standard energy and force labels to predict polarization-sensitive observables.
Long-range electrostatics for machine learning interatomic potentials is easier than we thought
Dec 19, 2025The lack of long-range electrostatics is a key limitation of modern machine learning interatomic potentials (MLIPs), hindering reliable applications to interfaces, charge-transfer reactions, polar and ionic materials, and biomolecules. In this Perspective, we distill two design principles behind the Latent Ewald Summation (LES) framework, which can capture long-range interactions, charges, and electrical response just by learning from standard energy and force training data: (i) use a Coulomb functional form with environment-dependent charges to capture electrostatic interactions, and (ii) avoid explicit training on ambiguous density functional theory (DFT) partial charges. When both principles are satisfied, substantial flexibility remains: essentially any short-range MLIP can be augmented; charge equilibration schemes can be added when desired; dipoles and Born effective charges can be inferred or finetuned; and charge/spin-state embeddings or tensorial targets can be further incorporated. We also discuss current limitations and open challenges. Together, these minimal, physics-guided design rules suggest that incorporating long-range electrostatics into MLIPs is simpler and perhaps more broadly applicable than is commonly assumed.
Machine learning interatomic potential can infer electrical response
Apr 07, 2025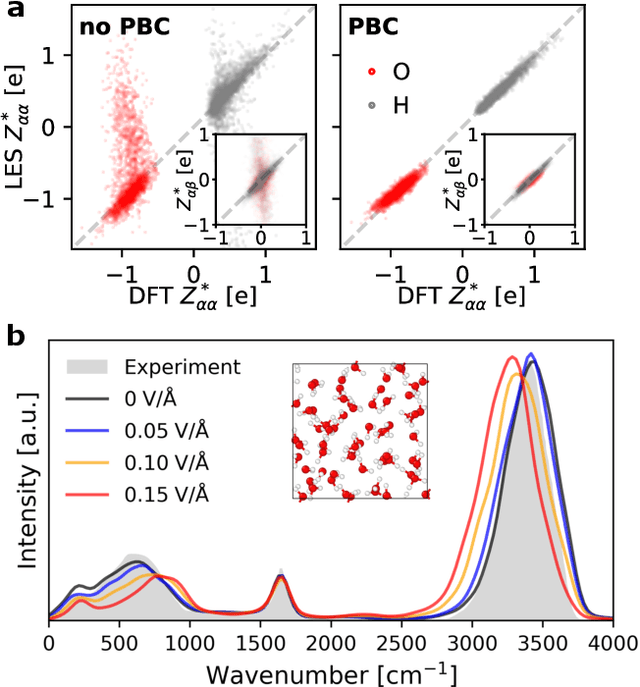
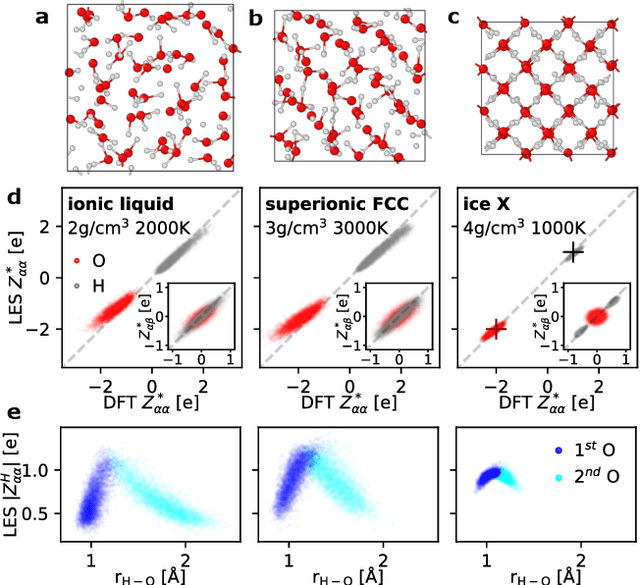
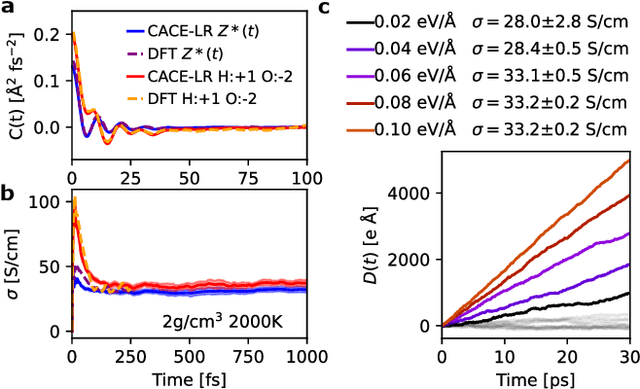
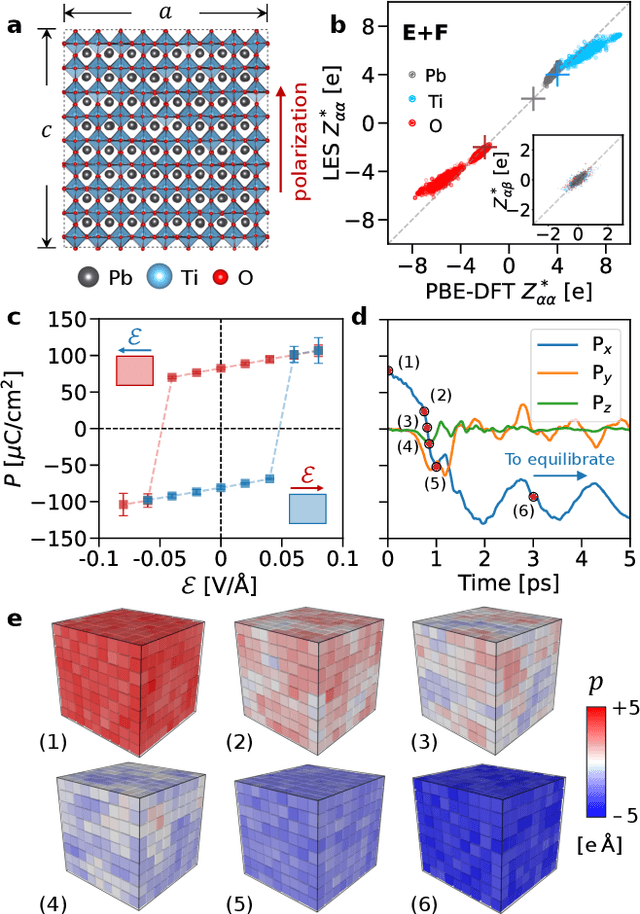
Modeling the response of material and chemical systems to electric fields remains a longstanding challenge. Machine learning interatomic potentials (MLIPs) offer an efficient and scalable alternative to quantum mechanical methods but do not by themselves incorporate electrical response. Here, we show that polarization and Born effective charge (BEC) tensors can be directly extracted from long-range MLIPs within the Latent Ewald Summation (LES) framework, solely by learning from energy and force data. Using this approach, we predict the infrared spectra of bulk water under zero or finite external electric fields, ionic conductivities of high-pressure superionic ice, and the phase transition and hysteresis in ferroelectric PbTiO$_3$ perovskite. This work thus extends the capability of MLIPs to predict electrical response--without training on charges or polarization or BECs--and enables accurate modeling of electric-field-driven processes in diverse systems at scale.
Learning charges and long-range interactions from energies and forces
Dec 19, 2024



Accurate modeling of long-range forces is critical in atomistic simulations, as they play a central role in determining the properties of materials and chemical systems. However, standard machine learning interatomic potentials (MLIPs) often rely on short-range approximations, limiting their applicability to systems with significant electrostatics and dispersion forces. We recently introduced the Latent Ewald Summation (LES) method, which captures long-range electrostatics without explicitly learning atomic charges or charge equilibration. Extending LES, we incorporate the ability to learn physical partial charges, encode charge states, and the option to impose charge neutrality constraints. We benchmark LES on diverse and challenging systems, including charged molecules, ionic liquid, electrolyte solution, polar dipeptides, surface adsorption, electrolyte/solid interfaces, and solid-solid interfaces. Our results show that LES can effectively infer physical partial charges, dipole and quadrupole moments, as well as achieve better accuracy compared to methods that explicitly learn charges. LES thus provides an efficient, interpretable, and generalizable MLIP framework for simulating complex systems with intricate charge transfer and long-range
Automatic feature selection and weighting using Differentiable Information Imbalance
Oct 30, 2024Feature selection is a common process in many applications, but it is accompanied by uncertainties such as: What is the optimal dimensionality of an interpretable, reduced feature space to retain a maximum amount of information? How to account for different units of measure in features? How to weight different features according to their importance? To address these challenges, we introduce the Differentiable Information Imbalance (DII), an automatic data analysis method to rank information content between sets of features. Based on the nearest neighbors according to distances in the ground truth feature space, the method finds a low-dimensional subset of the input features, within which the pairwise distance relations are most similar to the ground truth. By employing the Differentiable Information Imbalance as a loss function, the relative feature weights of the inputs are optimized, simultaneously performing unit alignment and relative importance scaling, while preserving interpretability. Furthermore, this method can generate sparse solutions and determine the optimal size of the reduced feature space. We illustrate the usefulness of this approach on two prototypical benchmark problems: (1) Identifying a small set of collective variables capable of describing the conformational space of a biomolecule, and (2) selecting a subset of features for training a machine-learning force field. The results highlight the potential of the Differentiable Information Imbalance in addressing feature selection challenges and optimizing dimensionality in various applications. The method is implemented in the Python library DADApy.
Latent Ewald summation for machine learning of long-range interactions
Aug 27, 2024



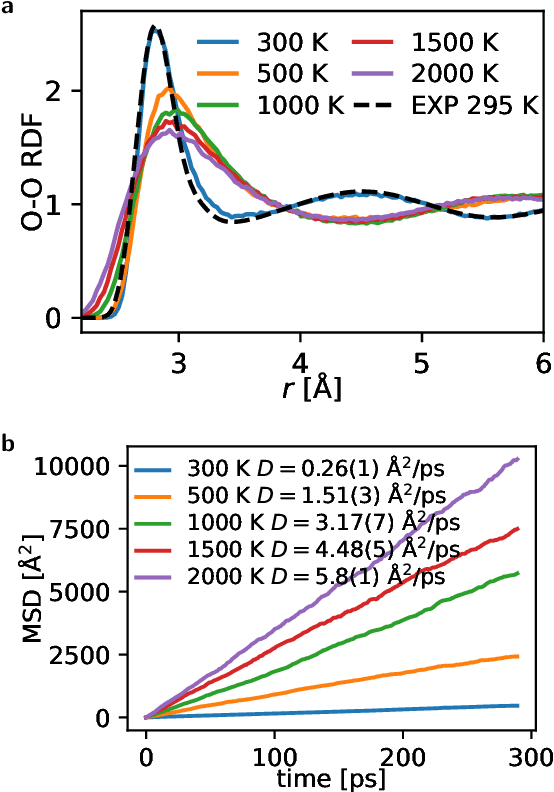
Machine learning interatomic potentials (MLIPs) often neglect long-range interactions, such as electrostatic and dispersion forces. In this work, we introduce a straightforward and efficient method to account for long-range interactions by learning a latent variable from local atomic descriptors and applying an Ewald summation to this variable. We demonstrate that in systems including charged, polar, or apolar molecular dimers, bulk water, and water-vapor interface, standard short-ranged MLIPs can lead to unphysical predictions even when employing message passing. The long-range models effectively eliminate these artifacts, with only about twice the computational cost of short-range MLIPs.
Response Matching for generating materials and molecules
May 15, 2024
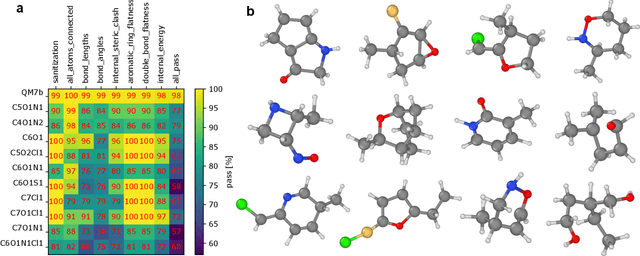

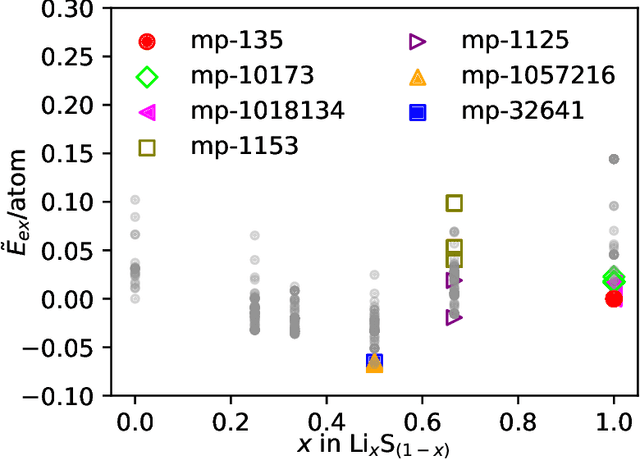
Machine learning has recently emerged as a powerful tool for generating new molecular and material structures. The success of state-of-the-art models stems from their ability to incorporate physical symmetries, such as translation, rotation, and periodicity. Here, we present a novel generative method called Response Matching (RM), which leverages the fact that each stable material or molecule exists at the minimum of its potential energy surface. Consequently, any perturbation induces a response in energy and stress, driving the structure back to equilibrium. Matching to such response is closely related to score matching in diffusion models. By employing the combination of a machine learning interatomic potential and random structure search as the denoising model, RM exploits the locality of atomic interactions, and inherently respects permutation, translation, rotation, and periodic invariances. RM is the first model to handle both molecules and bulk materials under the same framework. We demonstrate the efficiency and generalization of RM across three systems: a small organic molecular dataset, stable crystals from the Materials Project, and one-shot learning on a single diamond configuration.
Cartesian atomic cluster expansion for machine learning interatomic potentials
Feb 12, 2024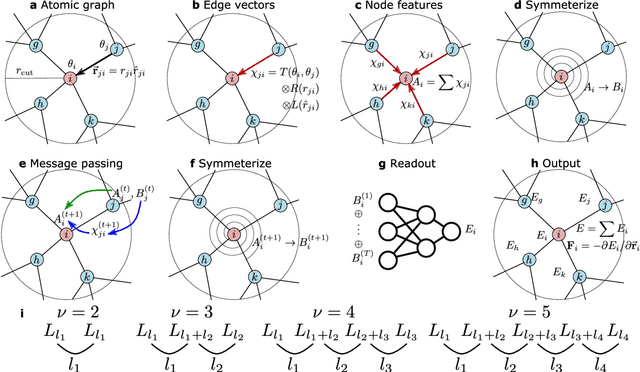
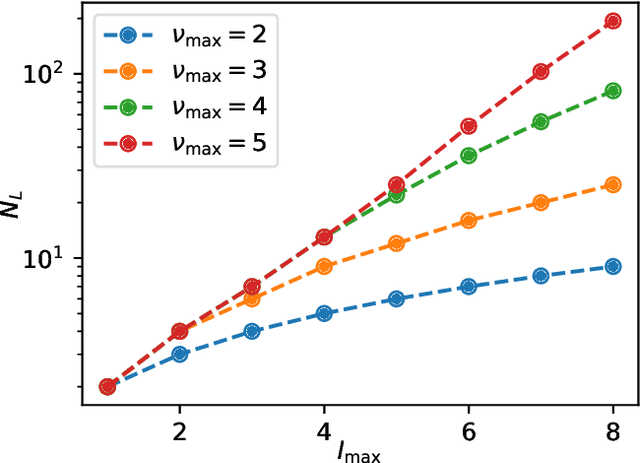
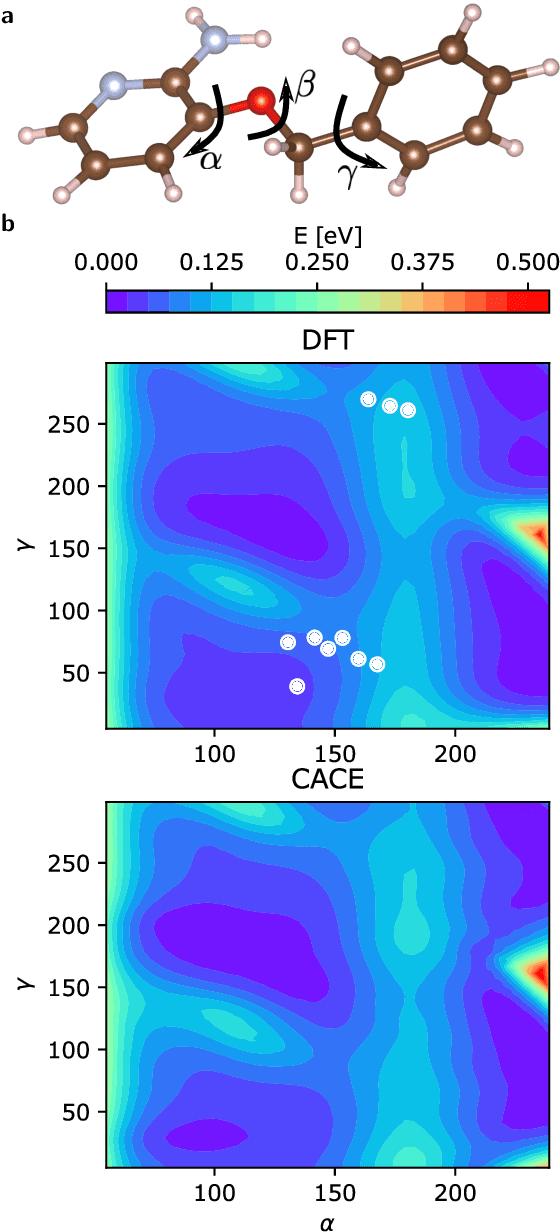
Machine learning interatomic potentials are revolutionizing large-scale, accurate atomistic modelling in material science and chemistry. These potentials often use atomic cluster expansion or equivariant message passing with spherical harmonics as basis functions. However, the dependence on Clebsch-Gordan coefficients for maintaining rotational symmetry leads to computational inefficiencies and redundancies. We propose an alternative: a Cartesian-coordinates-based atomic density expansion. This approach provides a complete description of atomic environments while maintaining interaction body orders. Additionally, we integrate low-dimensional embeddings of various chemical elements and inter-atomic message passing. The resulting potential, named Cartesian Atomic Cluster Expansion (CACE), exhibits good accuracy, stability, and generalizability. We validate its performance in diverse systems, including bulk water, small molecules, and 25-element high-entropy alloys.
BenchML: an extensible pipelining framework for benchmarking representations of materials and molecules at scale
Dec 04, 2021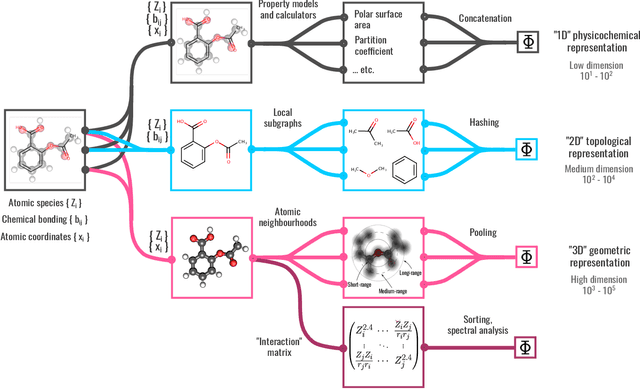
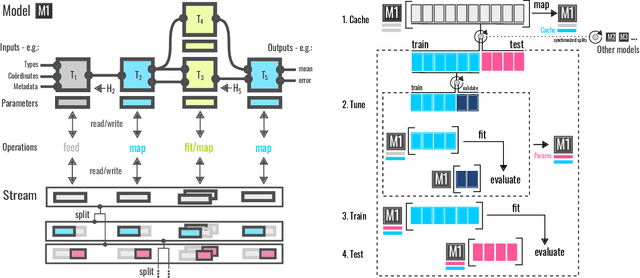
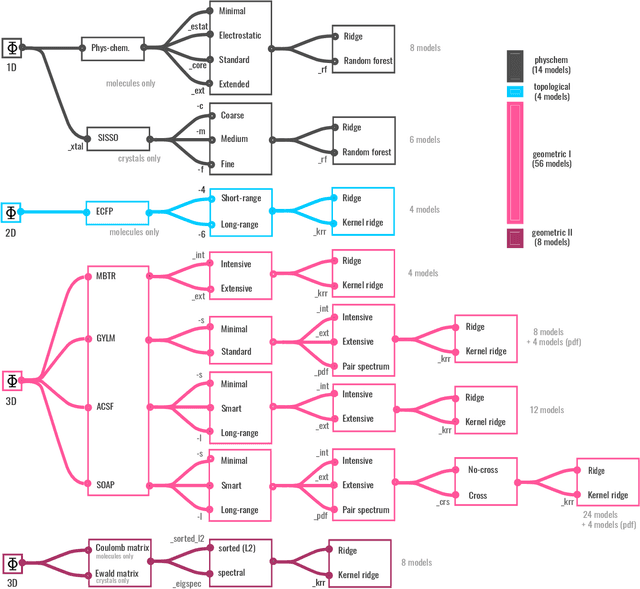
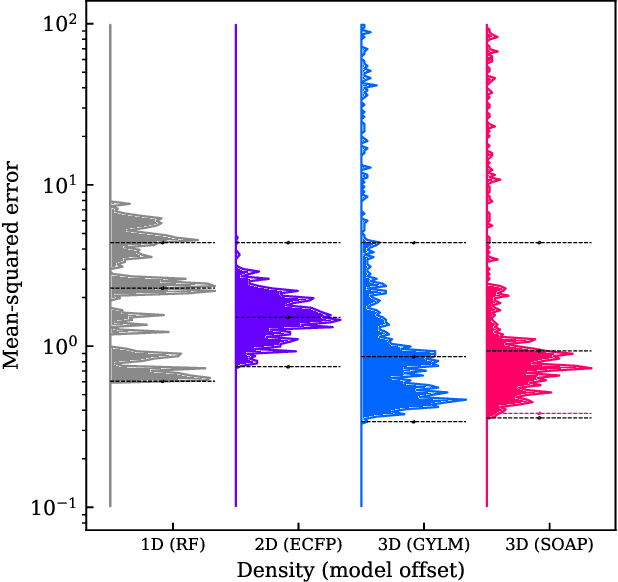
We introduce a machine-learning (ML) framework for high-throughput benchmarking of diverse representations of chemical systems against datasets of materials and molecules. The guiding principle underlying the benchmarking approach is to evaluate raw descriptor performance by limiting model complexity to simple regression schemes while enforcing best ML practices, allowing for unbiased hyperparameter optimization, and assessing learning progress through learning curves along series of synchronized train-test splits. The resulting models are intended as baselines that can inform future method development, next to indicating how easily a given dataset can be learnt. Through a comparative analysis of the training outcome across a diverse set of physicochemical, topological and geometric representations, we glean insight into the relative merits of these representations as well as their interrelatedness.