 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCartesian atomic cluster expansion for machine learning interatomic potentials
Paper and Code
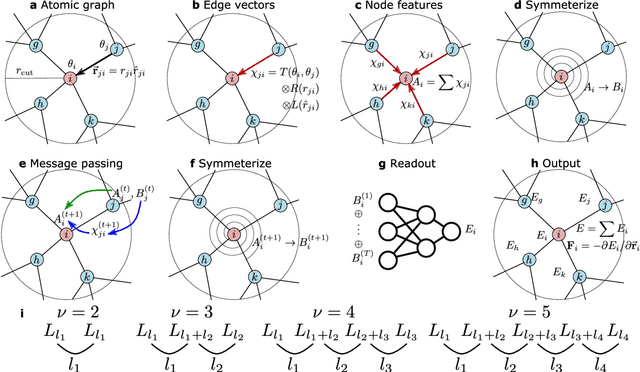
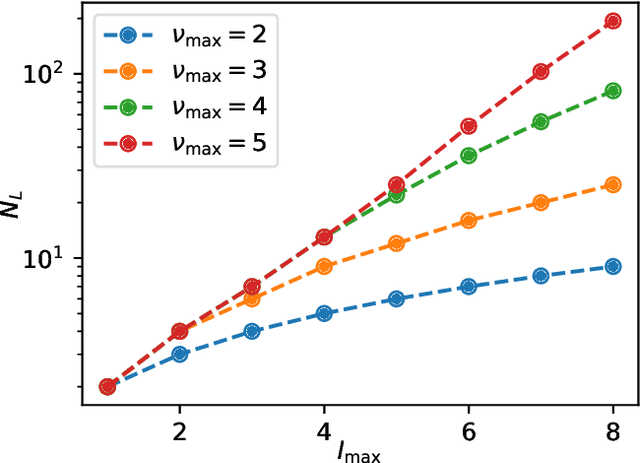
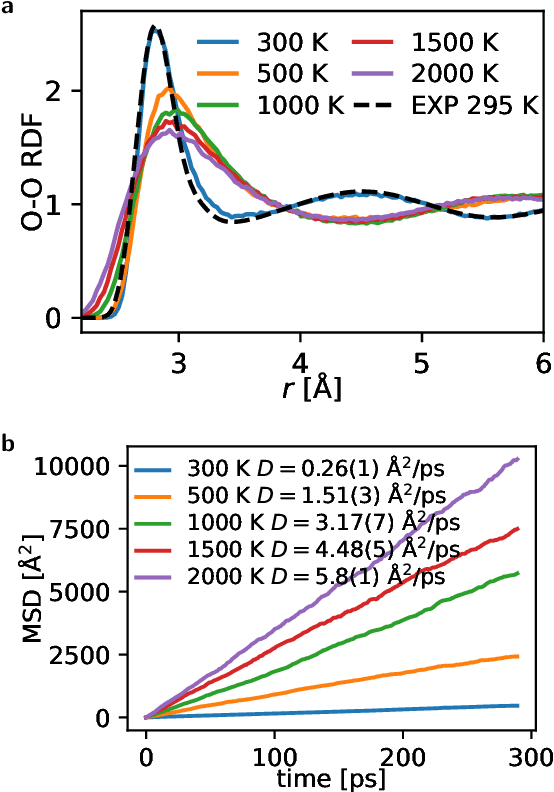
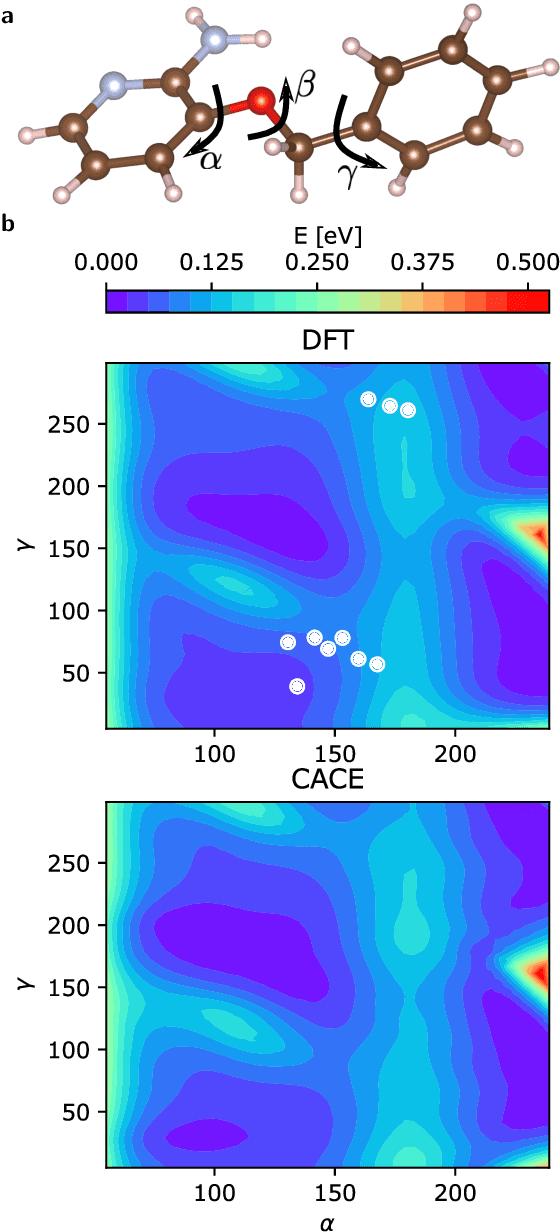
Machine learning interatomic potentials are revolutionizing large-scale, accurate atomistic modelling in material science and chemistry. These potentials often use atomic cluster expansion or equivariant message passing with spherical harmonics as basis functions. However, the dependence on Clebsch-Gordan coefficients for maintaining rotational symmetry leads to computational inefficiencies and redundancies. We propose an alternative: a Cartesian-coordinates-based atomic density expansion. This approach provides a complete description of atomic environments while maintaining interaction body orders. Additionally, we integrate low-dimensional embeddings of various chemical elements and inter-atomic message passing. The resulting potential, named Cartesian Atomic Cluster Expansion (CACE), exhibits good accuracy, stability, and generalizability. We validate its performance in diverse systems, including bulk water, small molecules, and 25-element high-entropy alloys.