 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOptimizing Data Distribution and Kernel Performance for Efficient Training of Chemistry Foundation Models: A Case Study with MACE
Apr 14, 2025



Chemistry Foundation Models (CFMs) that leverage Graph Neural Networks (GNNs) operating on 3D molecular graph structures are becoming indispensable tools for computational chemists and materials scientists. These models facilitate the understanding of matter and the discovery of new molecules and materials. In contrast to GNNs operating on a large homogeneous graphs, GNNs used by CFMs process a large number of geometric graphs of varying sizes, requiring different optimization strategies than those developed for large homogeneous GNNs. This paper presents optimizations for two critical phases of CFM training: data distribution and model training, targeting MACE - a state-of-the-art CFM. We address the challenge of load balancing in data distribution by formulating it as a multi-objective bin packing problem. We propose an iterative algorithm that provides a highly effective, fast, and practical solution, ensuring efficient data distribution. For the training phase, we identify symmetric tensor contraction as the key computational kernel in MACE and optimize this kernel to improve the overall performance. Our combined approach of balanced data distribution and kernel optimization significantly enhances the training process of MACE. Experimental results demonstrate a substantial speedup, reducing per-epoch execution time for training from 12 to 2 minutes on 740 GPUs with a 2.6M sample dataset.
A practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025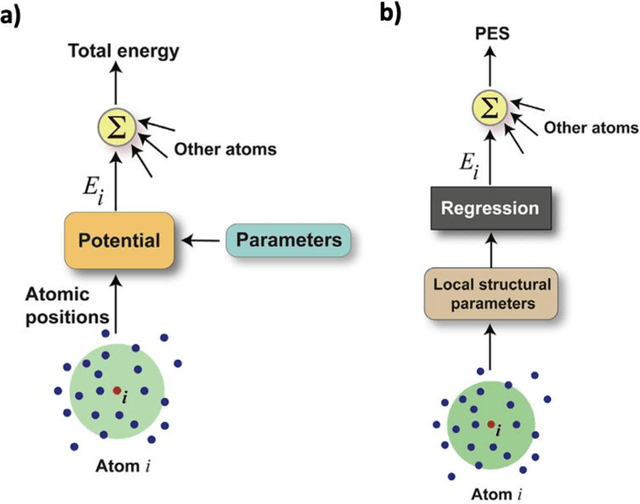

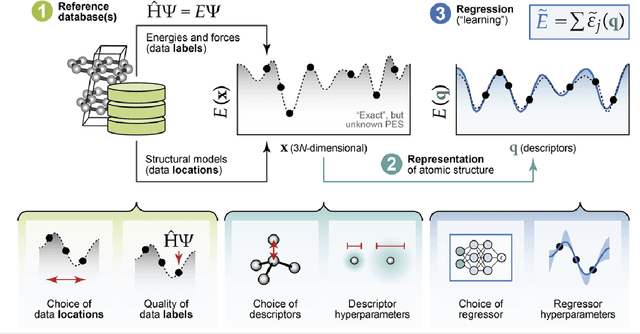
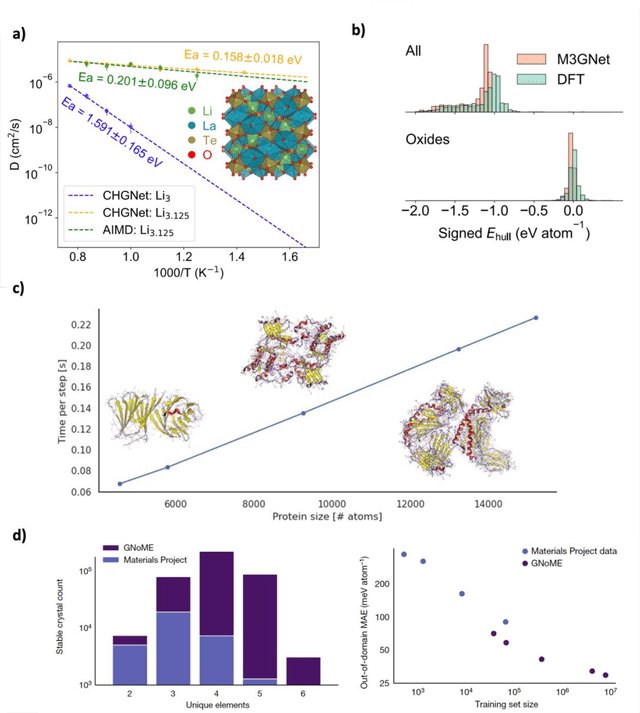
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
Evaluation of the MACE Force Field Architecture: from Medicinal Chemistry to Materials Science
May 23, 2023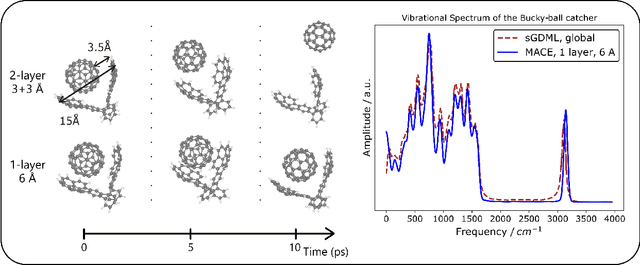
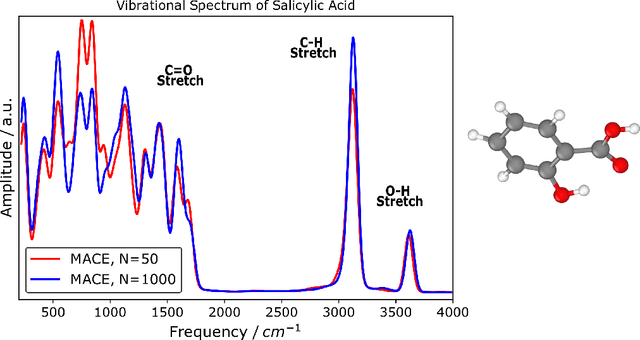
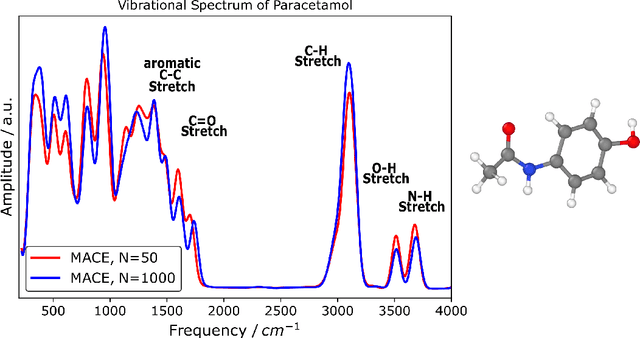
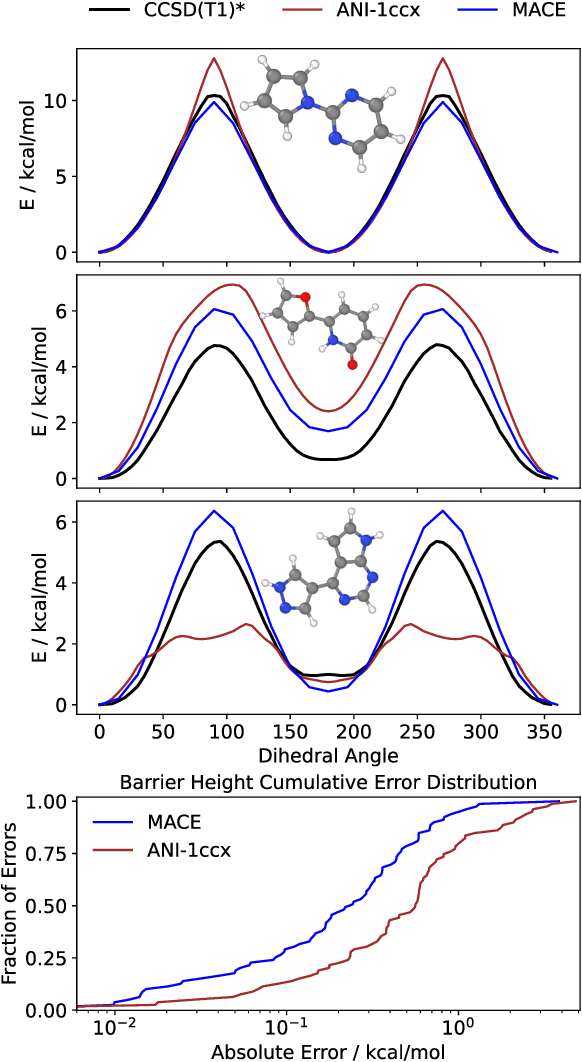
The MACE architecture represents the state of the art in the field of machine learning force fields for a variety of in-domain, extrapolation and low-data regime tasks. In this paper, we further evaluate MACE by fitting models for published benchmark datasets. We show that MACE generally outperforms alternatives for a wide range of systems from amorphous carbon and general small molecule organic chemistry to large molecules and liquid water. We demonstrate the capabilities of the model on tasks ranging from constrained geometry optimisation to molecular dynamics simulations and find excellent performance across all tested domains. We show that MACE is very data efficient, and can reproduce experimental molecular vibrational spectra when trained on as few as 50 randomly selected reference configurations. We further demonstrate that the strictly local atom-centered model is sufficient for such tasks even in the case of large molecules and weakly interacting molecular assemblies.
Ranking the information content of distance measures
Apr 30, 2021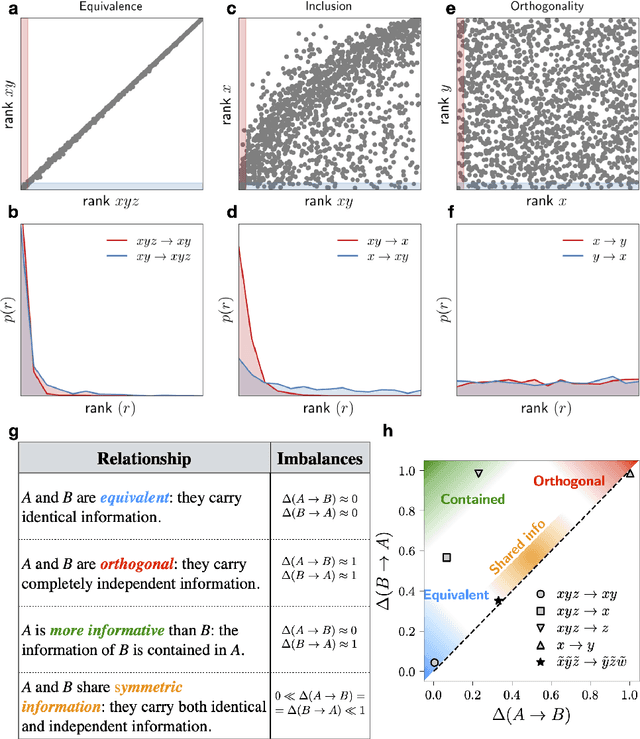
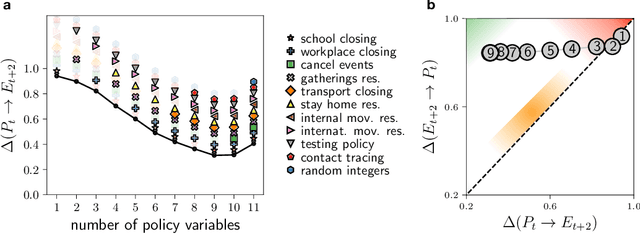
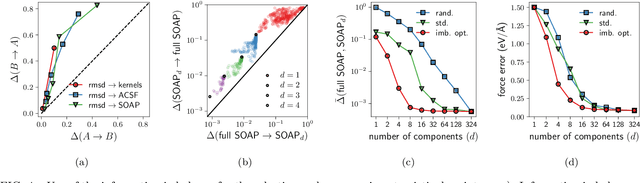
Real-world data typically contain a large number of features that are often heterogeneous in nature, relevance, and also units of measure. When assessing the similarity between data points, one can build various distance measures using subsets of these features. Using the fewest features but still retaining sufficient information about the system is crucial in many statistical learning approaches, particularly when data are sparse. We introduce a statistical test that can assess the relative information retained when using two different distance measures, and determine if they are equivalent, independent, or if one is more informative than the other. This in turn allows finding the most informative distance measure out of a pool of candidates. The approach is applied to find the most relevant policy variables for controlling the Covid-19 epidemic and to find compact yet informative representations of atomic structures, but its potential applications are wide ranging in many branches of science.
An Experimentally Driven Automated Machine Learned lnter-Atomic Potential for a Refractory Oxide
Sep 09, 2020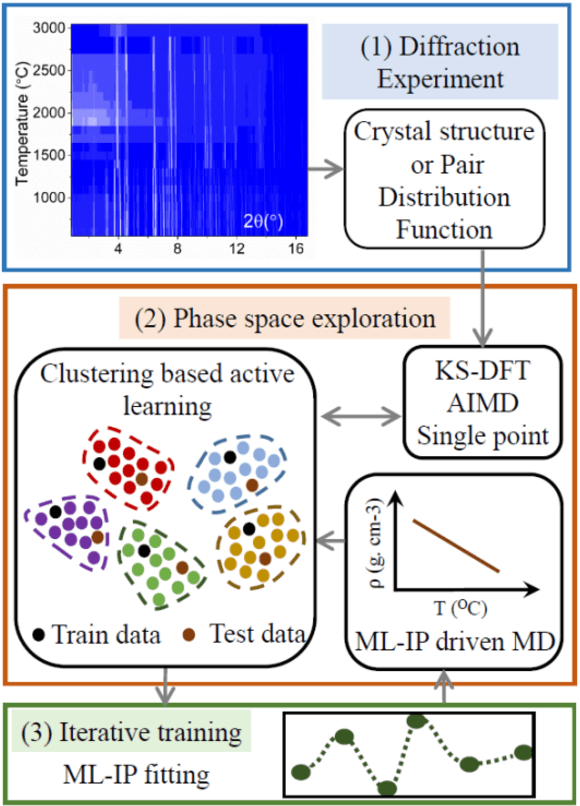
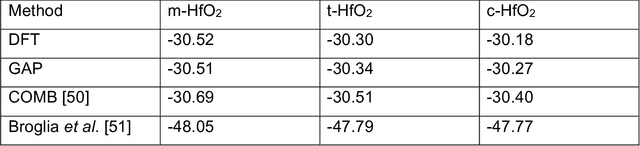
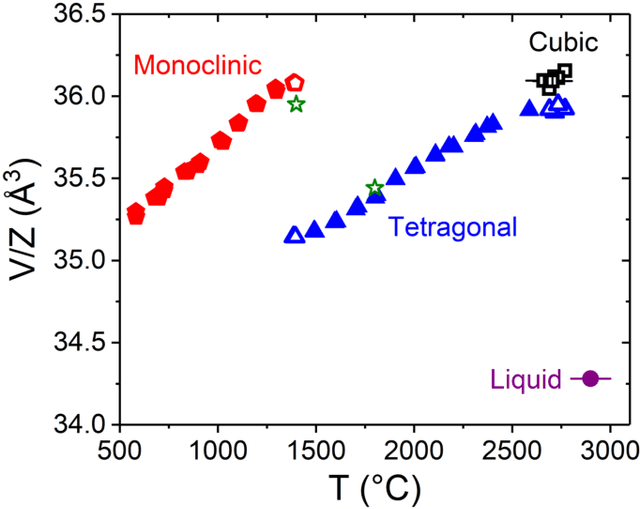
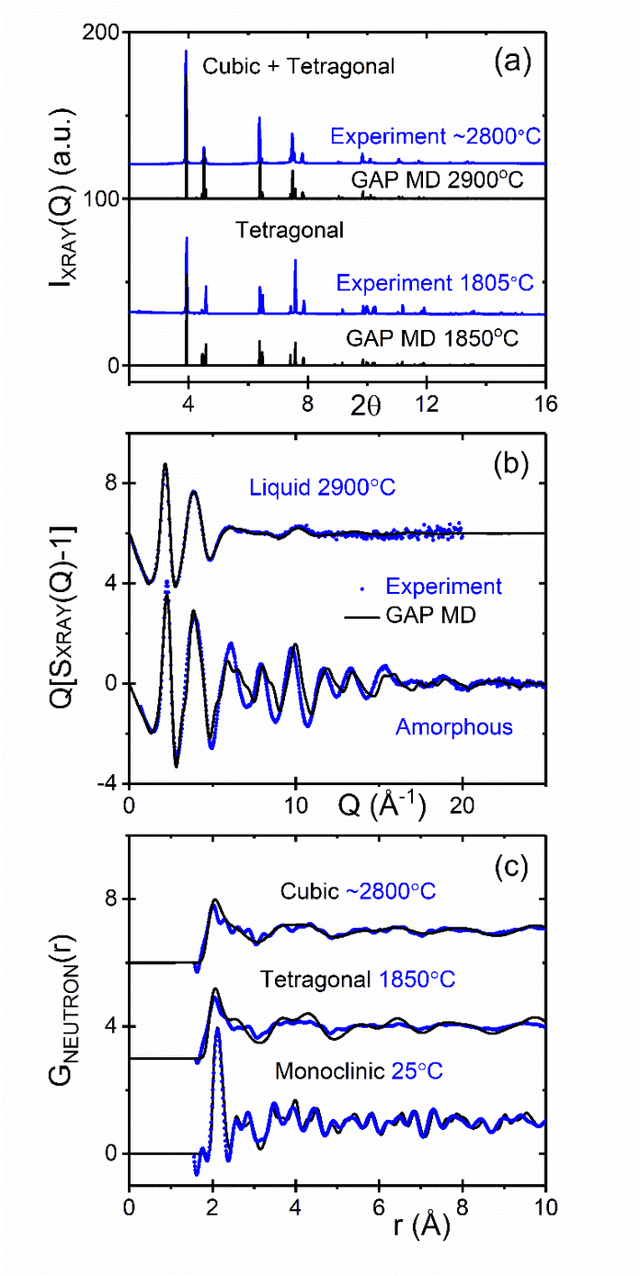
Understanding the structure and properties of refractory oxides are critical for high temperature applications. In this work, a combined experimental and simulation approach uses an automated closed loop via an active-learner, which is initialized by X-ray and neutron diffraction measurements, and sequentially improves a machine-learning model until the experimentally predetermined phase space is covered. A multi-phase potential is generated for a canonical example of the archetypal refractory oxide, HfO2, by drawing a minimum number of training configurations from room temperature to the liquid state at ~2900oC. The method significantly reduces model development time and human effort.