 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNoisy, sparse, nonlinear: Navigating the Bermuda Triangle of physical inference with deep filtering
Nov 19, 2019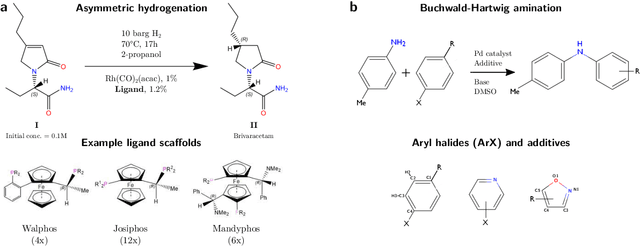
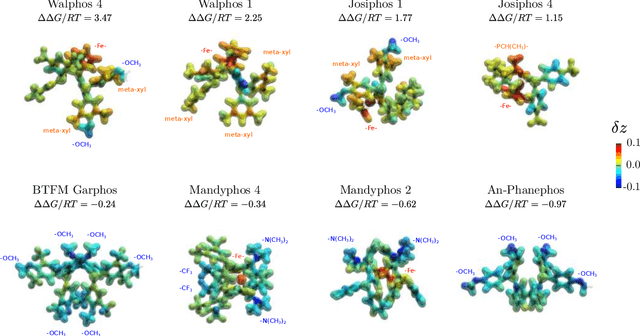
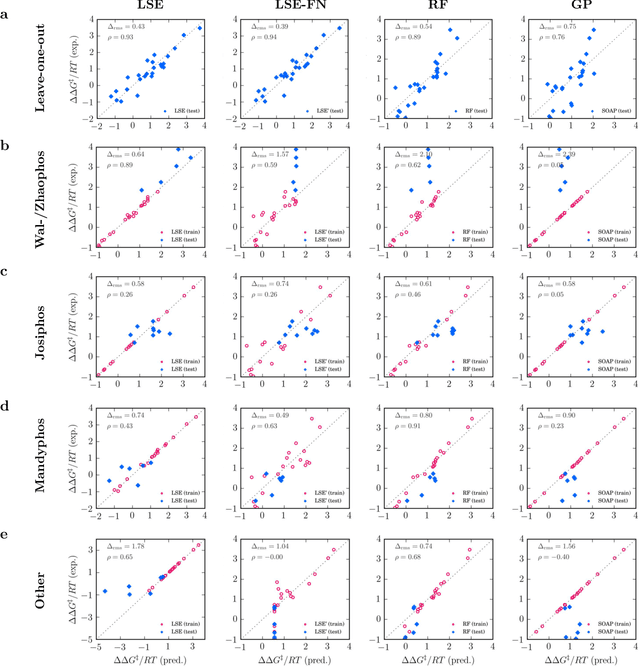
Capturing the microscopic interactions that determine molecular reactivity poses a challenge across the physical sciences. Even a basic understanding of the underlying reaction mechanisms can substantially accelerate materials and compound design, including the development of new catalysts or drugs. Given the difficulties routinely faced by both experimental and theoretical investigations that aim to improve our mechanistic understanding of a reaction, recent advances have focused on data-driven routes to derive structure-property relationships directly from high-throughput screens. However, even these high-quality, high-volume data are noisy, sparse and biased -- placing them in a regime where machine-learning is extremely challenging. Here we show that a statistical approach based on deep filtering of nonlinear feature networks results in physicochemical models that are more robust, transparent and generalize better than standard machine-learning architectures. Using diligent descriptor design and data post-processing, we exemplify the approach using both literature and fresh data on asymmetric catalytic hydrogenation, Palladium-catalyzed cross-coupling reactions, and drug-drug synergy. We illustrate how the sparse models uncovered by the filtering help us formulate physicochemical reaction ``pharmacophores'', investigate experimental bias and derive strategies for mechanism detection and classification.