 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOpenContrails: Benchmarking Contrail Detection on GOES-16 ABI
Apr 20, 2023



Contrails (condensation trails) are line-shaped ice clouds caused by aircraft and are likely the largest contributor of aviation-induced climate change. Contrail avoidance is potentially an inexpensive way to significantly reduce the climate impact of aviation. An automated contrail detection system is an essential tool to develop and evaluate contrail avoidance systems. In this paper, we present a human-labeled dataset named OpenContrails to train and evaluate contrail detection models based on GOES-16 Advanced Baseline Imager (ABI) data. We propose and evaluate a contrail detection model that incorporates temporal context for improved detection accuracy. The human labeled dataset and the contrail detection outputs are publicly available on Google Cloud Storage at gs://goes_contrails_dataset.
Crystal Structure Search with Random Relaxations Using Graph Networks
Dec 08, 2020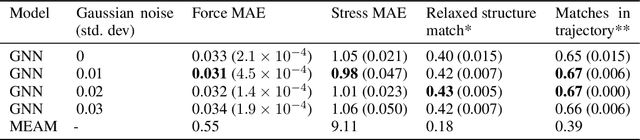
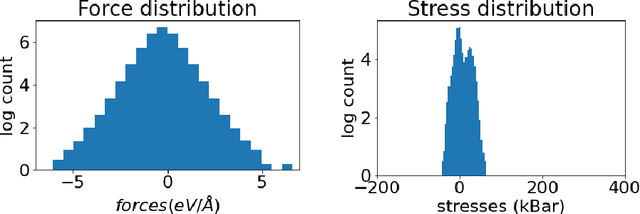
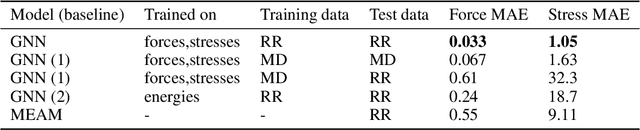
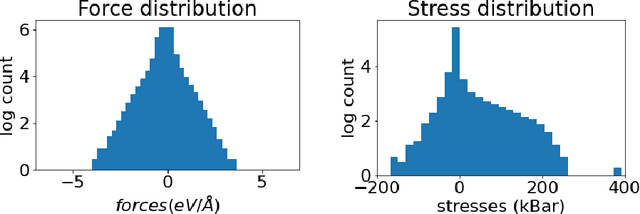
Materials design enables technologies critical to humanity, including combating climate change with solar cells and batteries. Many properties of a material are determined by its atomic crystal structure. However, prediction of the atomic crystal structure for a given material's chemical formula is a long-standing grand challenge that remains a barrier in materials design. We investigate a data-driven approach to accelerating ab initio random structure search (AIRSS), a state-of-the-art method for crystal structure search. We build a novel dataset of random structure relaxations of Li-Si battery anode materials using high-throughput density functional theory calculations. We train graph neural networks to simulate relaxations of random structures. Our model is able to find an experimentally verified structure of Li15Si4 it was not trained on, and has potential for orders of magnitude speedup over AIRSS when searching large unit cells and searching over multiple chemical stoichiometries. Surprisingly, we find that data augmentation of adding Gaussian noise improves both the accuracy and out of domain generalization of our models.
Machine learning on DNA-encoded libraries: A new paradigm for hit-finding
Jan 31, 2020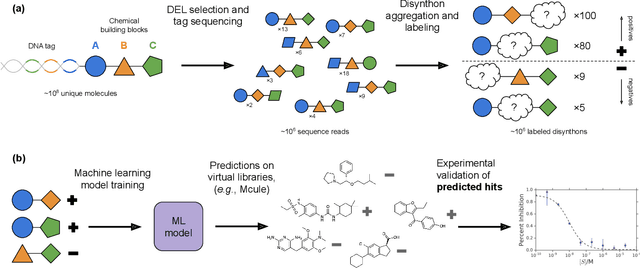
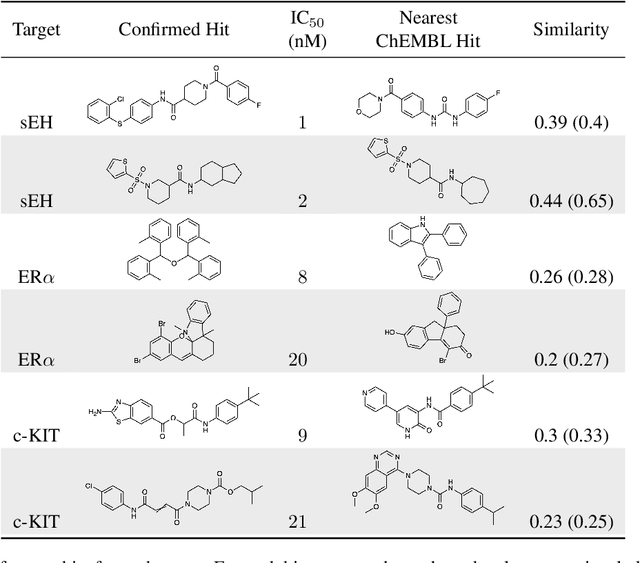
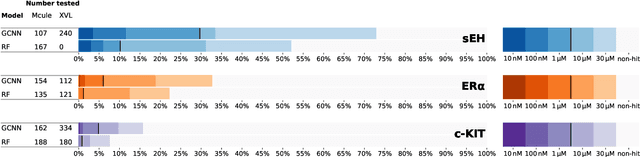
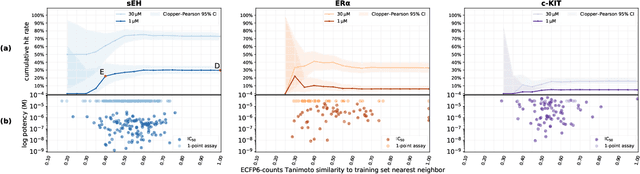
DNA-encoded small molecule libraries (DELs) have enabled discovery of novel inhibitors for many distinct protein targets of therapeutic value through screening of libraries with up to billions of unique small molecules. We demonstrate a new approach applying machine learning to DEL selection data by identifying active molecules from a large commercial collection and a virtual library of easily synthesizable compounds. We train models using only DEL selection data and apply automated or automatable filters with chemist review restricted to the removal of molecules with potential for instability or reactivity. We validate this approach with a large prospective study (nearly 2000 compounds tested) across three diverse protein targets: sEH (a hydrolase), ER{\alpha} (a nuclear receptor), and c-KIT (a kinase). The approach is effective, with an overall hit rate of {\sim}30% at 30 {\textmu}M and discovery of potent compounds (IC50 <10 nM) for every target. The model makes useful predictions even for molecules dissimilar to the original DEL and the compounds identified are diverse, predominantly drug-like, and different from known ligands. Collectively, the quality and quantity of DEL selection data; the power of modern machine learning methods; and access to large, inexpensive, commercially-available libraries creates a powerful new approach for hit finding.
Using Attribution to Decode Dataset Bias in Neural Network Models for Chemistry
Nov 29, 2018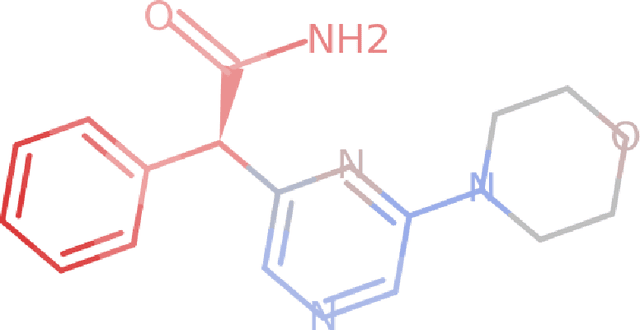
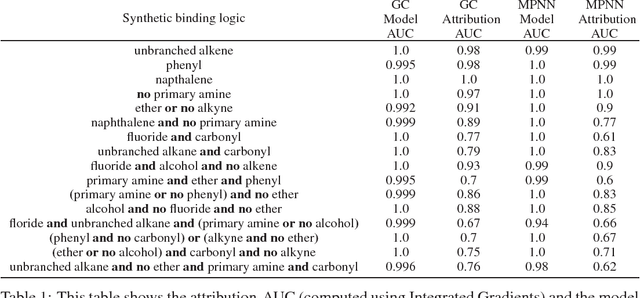
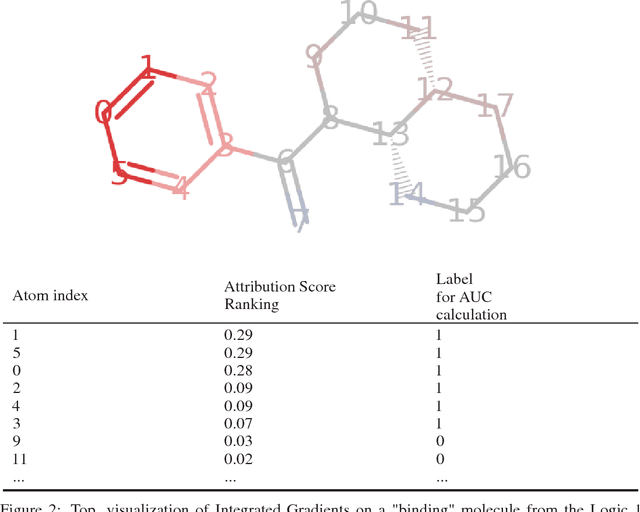
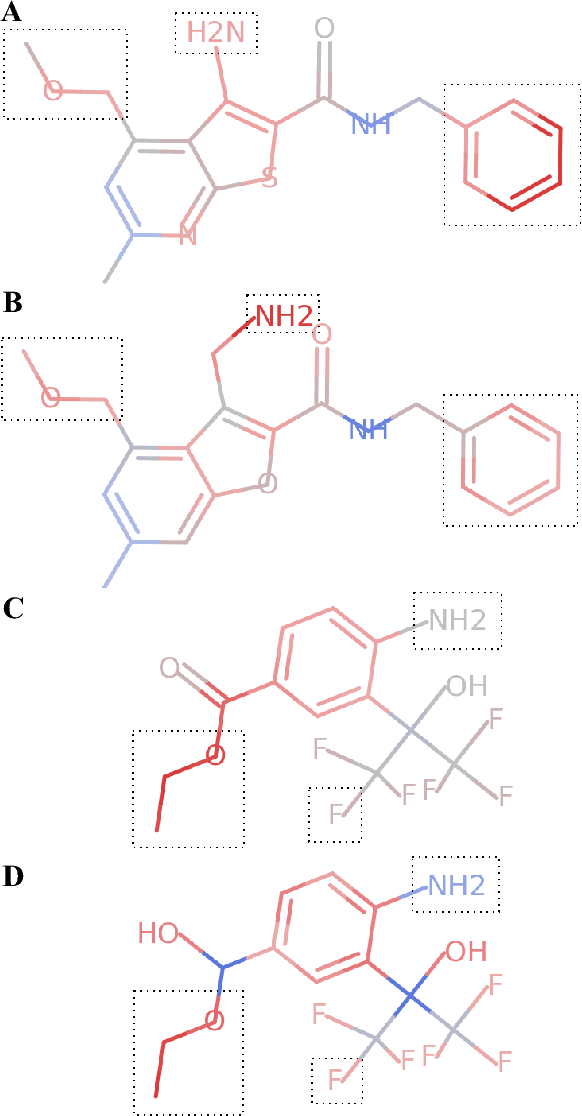
Deep neural networks have achieved state of the art accuracy at classifying molecules with respect to whether they bind to specific protein targets. A key breakthrough would occur if these models could reveal the fragment pharmacophores that are causally involved in binding. Extracting chemical details of binding from the networks could potentially lead to scientific discoveries about the mechanisms of drug actions. But doing so requires shining light into the black box that is the trained neural network model, a task that has proved difficult across many domains. Here we show how the binding mechanism learned by deep neural network models can be interrogated, using a recently described attribution method. We first work with carefully constructed synthetic datasets, in which the 'fragment logic' of binding is fully known. We find that networks that achieve perfect accuracy on held out test datasets still learn spurious correlations due to biases in the datasets, and we are able to exploit this non-robustness to construct adversarial examples that fool the model. The dataset bias makes these models unreliable for accurately revealing information about the mechanisms of protein-ligand binding. In light of our findings, we prescribe a test that checks for dataset bias given a hypothesis. If the test fails, it indicates that either the model must be simplified or regularized and/or that the training dataset requires augmentation.
Molecular Graph Convolutions: Moving Beyond Fingerprints
Aug 18, 2016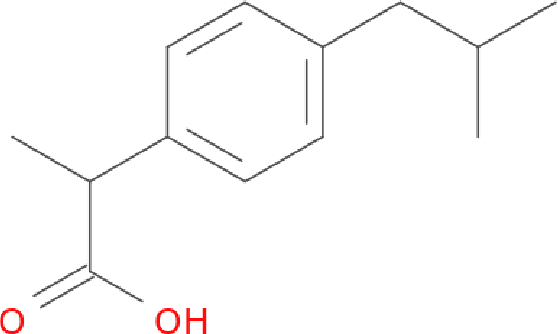
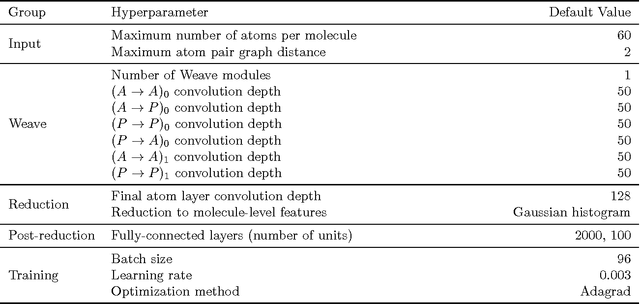
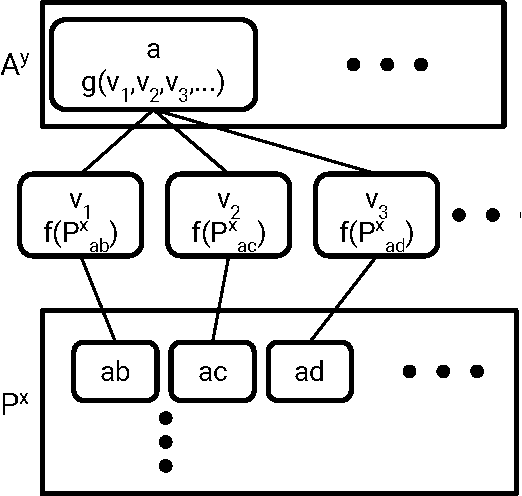
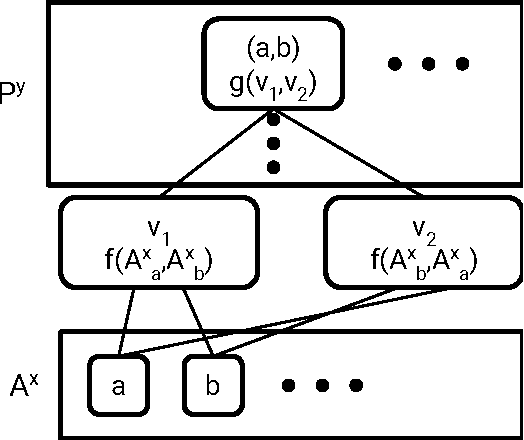
Molecular "fingerprints" encoding structural information are the workhorse of cheminformatics and machine learning in drug discovery applications. However, fingerprint representations necessarily emphasize particular aspects of the molecular structure while ignoring others, rather than allowing the model to make data-driven decisions. We describe molecular "graph convolutions", a machine learning architecture for learning from undirected graphs, specifically small molecules. Graph convolutions use a simple encoding of the molecular graph---atoms, bonds, distances, etc.---which allows the model to take greater advantage of information in the graph structure. Although graph convolutions do not outperform all fingerprint-based methods, they (along with other graph-based methods) represent a new paradigm in ligand-based virtual screening with exciting opportunities for future improvement.
* See "Version information" section