 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePursuing a Prospective Perspective
Aug 26, 2020Retrospective testing of predictive models does not consider the real-world context in which models are deployed. Prospective validation, on the other hand, enables meaningful comparisons between data generation processes by incorporating trained models and considering the subjective decisions that affect reproducibility. Prospective experiments are essential for consistent progress in modeling.
Energy-based View of Retrosynthesis
Jul 14, 2020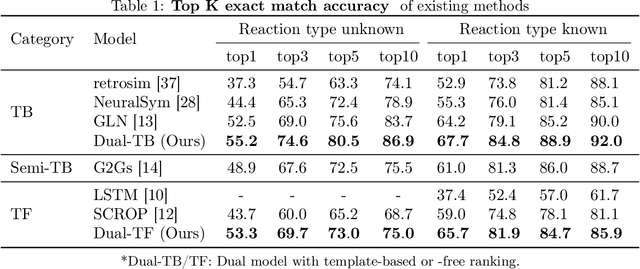
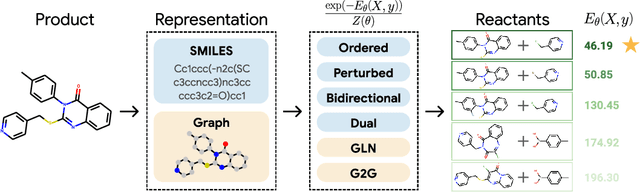
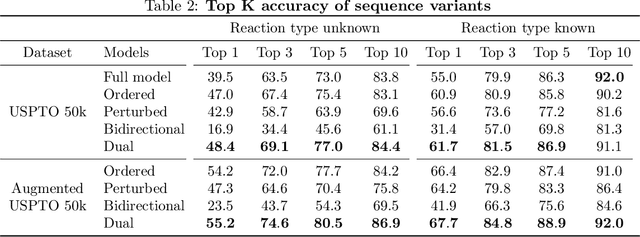

Retrosynthesis -- the process of identifying a set of reactants to synthesize a target molecule -- is of vital importance to material design and drug discovery. Existing machine learning approaches based on language models and graph neural networks have achieved encouraging results. In this paper, we propose a framework that unifies sequence- and graph-based methods as energy-based models (EBMs) with different energy functions. This unified perspective provides critical insights about EBM variants through a comprehensive assessment of performance. Additionally, we present a novel dual variant within the framework that performs consistent training over Bayesian forward- and backward-prediction by constraining the agreement between the two directions. This model improves state-of-the-art performance by 9.6% for template-free approaches where the reaction type is unknown.
Machine learning on DNA-encoded libraries: A new paradigm for hit-finding
Jan 31, 2020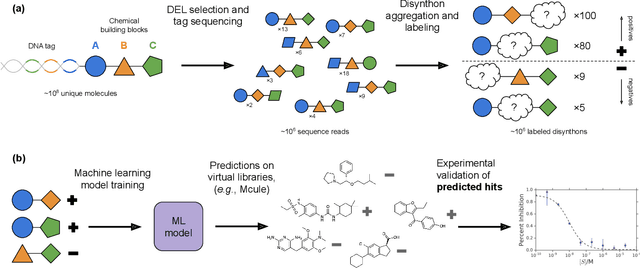
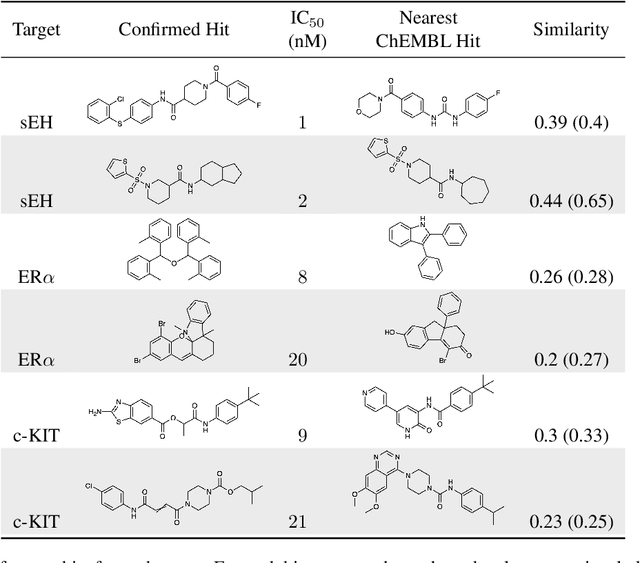
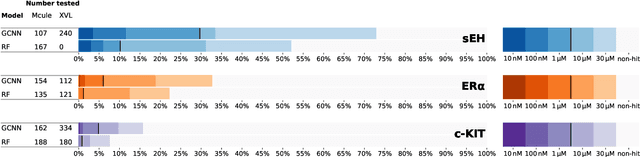
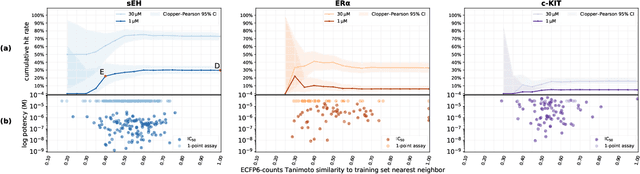
DNA-encoded small molecule libraries (DELs) have enabled discovery of novel inhibitors for many distinct protein targets of therapeutic value through screening of libraries with up to billions of unique small molecules. We demonstrate a new approach applying machine learning to DEL selection data by identifying active molecules from a large commercial collection and a virtual library of easily synthesizable compounds. We train models using only DEL selection data and apply automated or automatable filters with chemist review restricted to the removal of molecules with potential for instability or reactivity. We validate this approach with a large prospective study (nearly 2000 compounds tested) across three diverse protein targets: sEH (a hydrolase), ER{\alpha} (a nuclear receptor), and c-KIT (a kinase). The approach is effective, with an overall hit rate of {\sim}30% at 30 {\textmu}M and discovery of potent compounds (IC50 <10 nM) for every target. The model makes useful predictions even for molecules dissimilar to the original DEL and the compounds identified are diverse, predominantly drug-like, and different from known ligands. Collectively, the quality and quantity of DEL selection data; the power of modern machine learning methods; and access to large, inexpensive, commercially-available libraries creates a powerful new approach for hit finding.
Decoding Molecular Graph Embeddings with Reinforcement Learning
Apr 18, 2019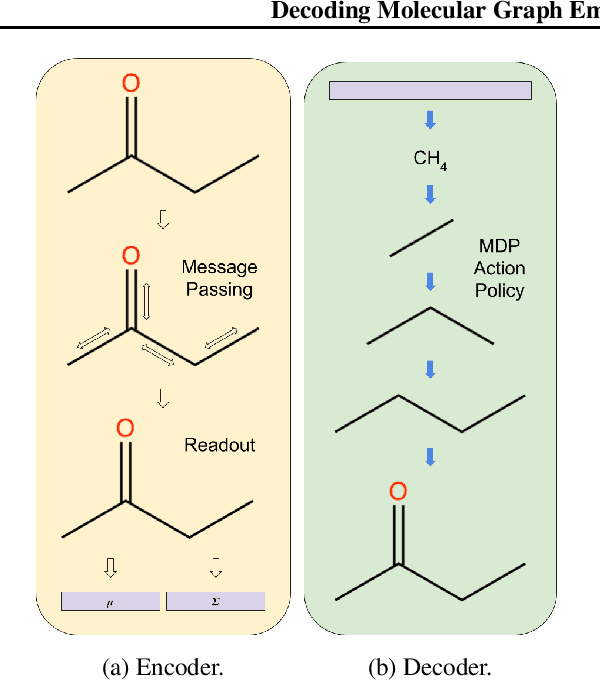
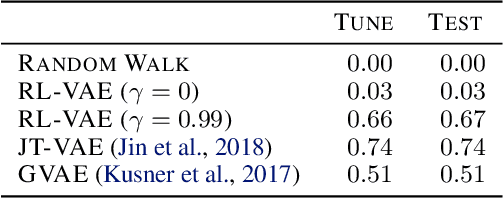
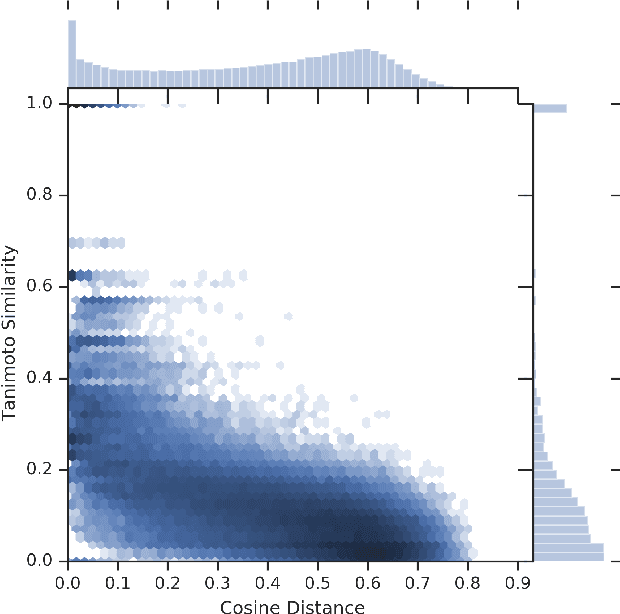
We present RL-VAE, a graph-to-graph variational autoencoder that uses reinforcement learning to decode molecular graphs from latent embeddings. Methods have been described previously for graph-to-graph autoencoding, but these approaches require sophisticated decoders that increase the complexity of training and evaluation (such as requiring parallel encoders and decoders or non-trivial graph matching). Here, we repurpose a simple graph generator to enable efficient decoding and generation of molecular graphs.
Optimization of Molecules via Deep Reinforcement Learning
Oct 23, 2018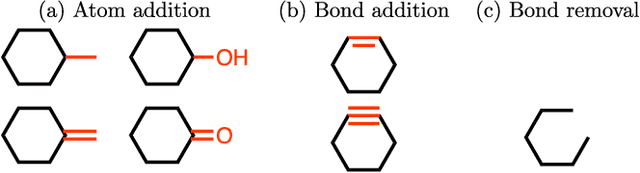
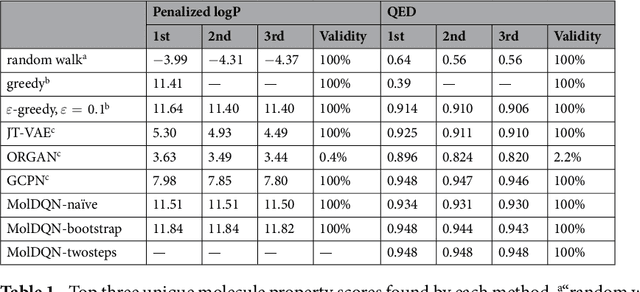
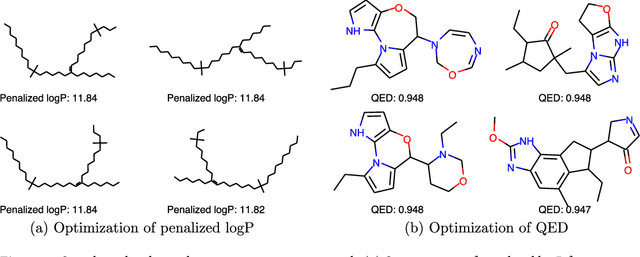
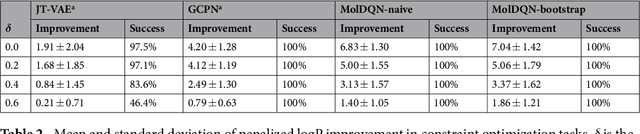
We present a framework, which we call Molecule Deep $Q$-Networks (MolDQN), for molecule optimization by combining domain knowledge of chemistry and state-of-the-art reinforcement learning techniques (prioritized experience replay, double $Q$-learning, and randomized value functions). We directly define modifications on molecules, thereby ensuring 100% chemical validity. Further, we operate without pre-training on any dataset to avoid possible bias from the choice of that set. As a result, our model outperforms several other state-of-the-art algorithms by having a higher success rate of acquiring molecules with better properties. Inspired by problems faced during medicinal chemistry lead optimization, we extend our model with multi-objective reinforcement learning, which maximizes drug-likeness while maintaining similarity to the original molecule. We further show the path through chemical space to achieve optimization for a molecule to understand how the model works.
Tensor field networks: Rotation- and translation-equivariant neural networks for 3D point clouds
May 18, 2018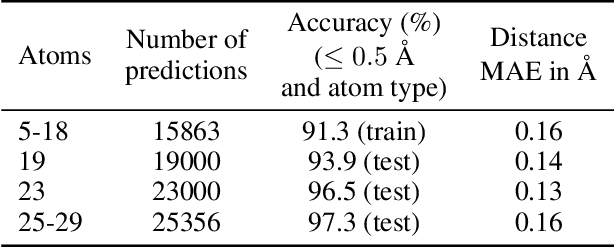
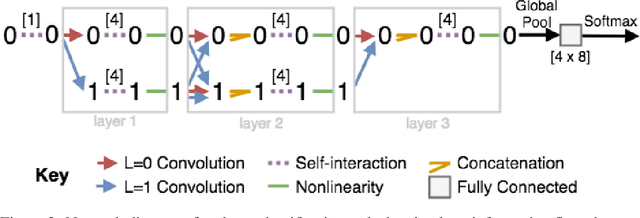
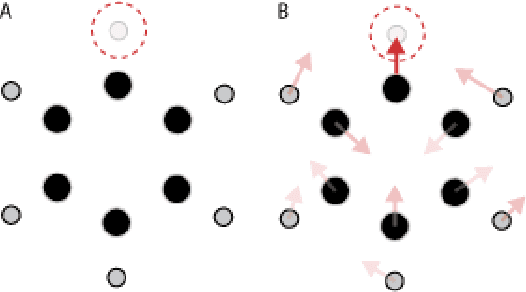
We introduce tensor field neural networks, which are locally equivariant to 3D rotations, translations, and permutations of points at every layer. 3D rotation equivariance removes the need for data augmentation to identify features in arbitrary orientations. Our network uses filters built from spherical harmonics; due to the mathematical consequences of this filter choice, each layer accepts as input (and guarantees as output) scalars, vectors, and higher-order tensors, in the geometric sense of these terms. We demonstrate the capabilities of tensor field networks with tasks in geometry, physics, and chemistry.
Modeling Industrial ADMET Data with Multitask Networks
Jan 13, 2017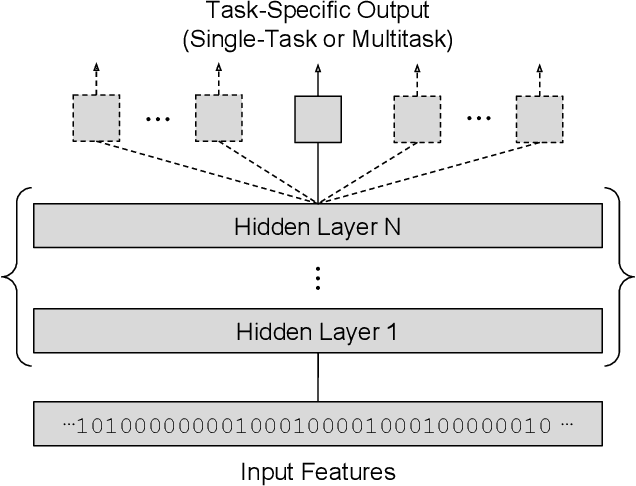
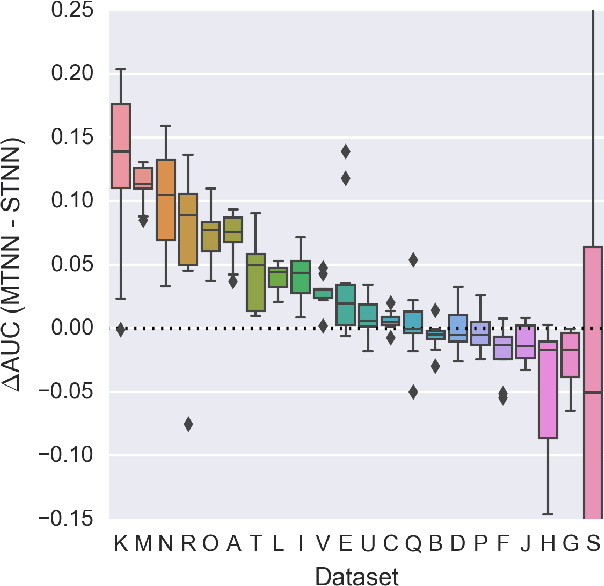
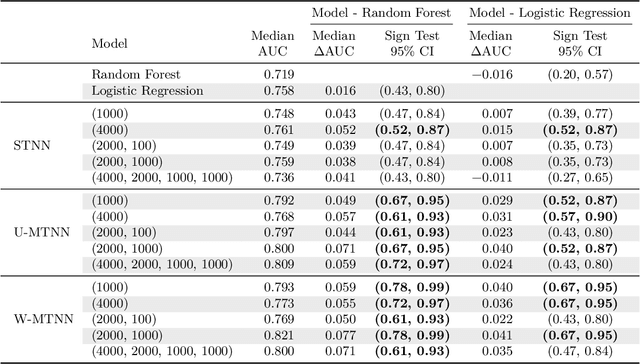
Deep learning methods such as multitask neural networks have recently been applied to ligand-based virtual screening and other drug discovery applications. Using a set of industrial ADMET datasets, we compare neural networks to standard baseline models and analyze multitask learning effects with both random cross-validation and a more relevant temporal validation scheme. We confirm that multitask learning can provide modest benefits over single-task models and show that smaller datasets tend to benefit more than larger datasets from multitask learning. Additionally, we find that adding massive amounts of side information is not guaranteed to improve performance relative to simpler multitask learning. Our results emphasize that multitask effects are highly dataset-dependent, suggesting the use of dataset-specific models to maximize overall performance.
ROCS-Derived Features for Virtual Screening
Aug 22, 2016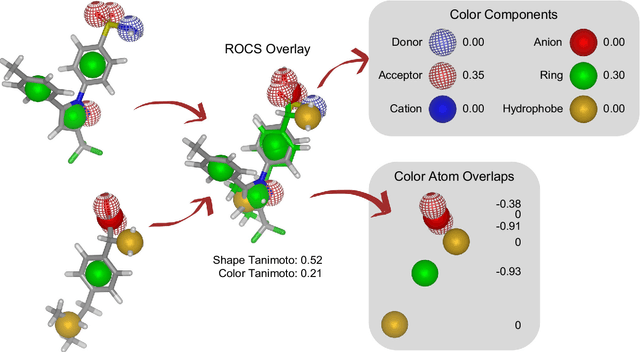

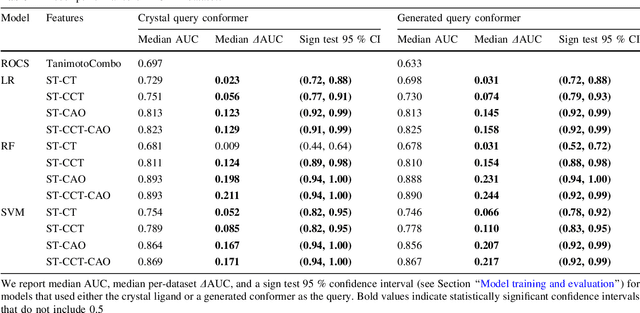
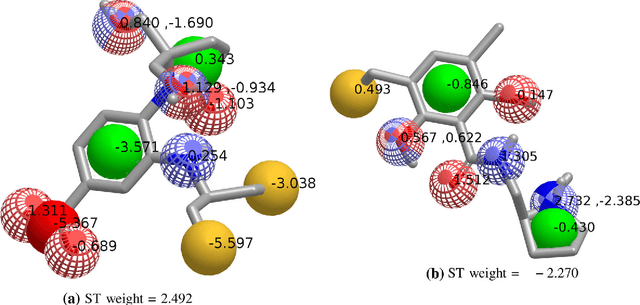
Rapid overlay of chemical structures (ROCS) is a standard tool for the calculation of 3D shape and chemical ("color") similarity. ROCS uses unweighted sums to combine many aspects of similarity, yielding parameter-free models for virtual screening. In this report, we decompose the ROCS color force field into "color components" and "color atom overlaps", novel color similarity features that can be weighted in a system-specific manner by machine learning algorithms. In cross-validation experiments, these additional features significantly improve virtual screening performance (ROC AUC scores) relative to standard ROCS.
Molecular Graph Convolutions: Moving Beyond Fingerprints
Aug 18, 2016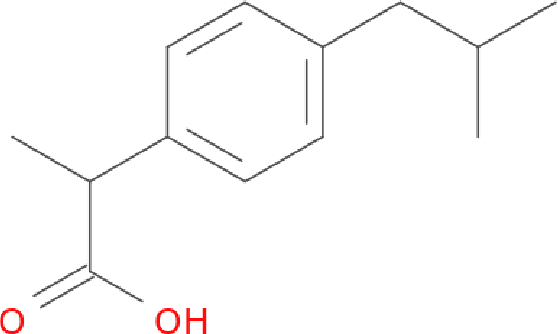
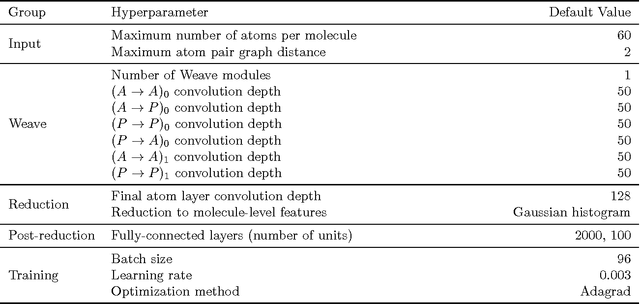
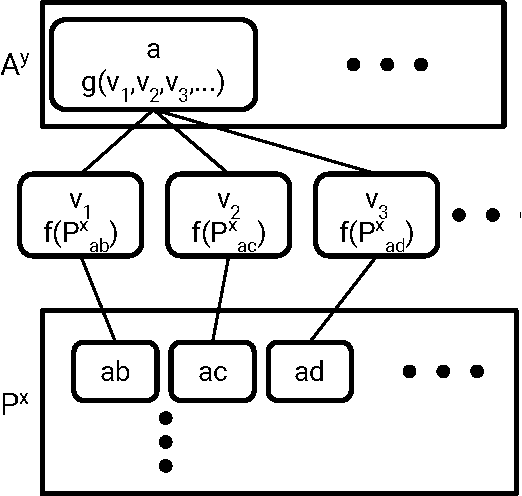
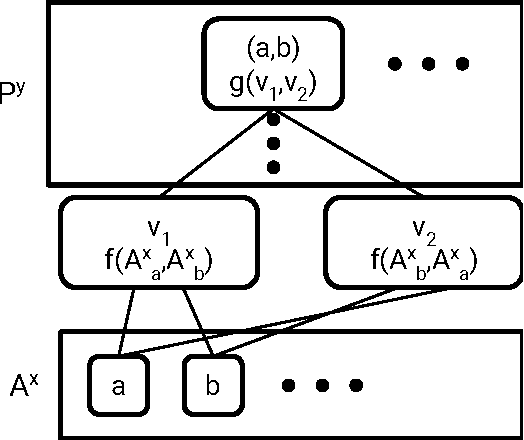
Molecular "fingerprints" encoding structural information are the workhorse of cheminformatics and machine learning in drug discovery applications. However, fingerprint representations necessarily emphasize particular aspects of the molecular structure while ignoring others, rather than allowing the model to make data-driven decisions. We describe molecular "graph convolutions", a machine learning architecture for learning from undirected graphs, specifically small molecules. Graph convolutions use a simple encoding of the molecular graph---atoms, bonds, distances, etc.---which allows the model to take greater advantage of information in the graph structure. Although graph convolutions do not outperform all fingerprint-based methods, they (along with other graph-based methods) represent a new paradigm in ligand-based virtual screening with exciting opportunities for future improvement.
* See "Version information" section
Massively Multitask Networks for Drug Discovery
Feb 06, 2015
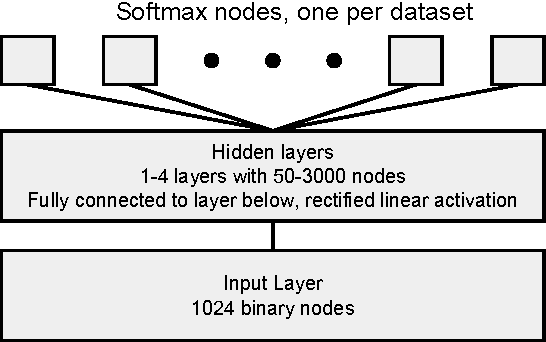
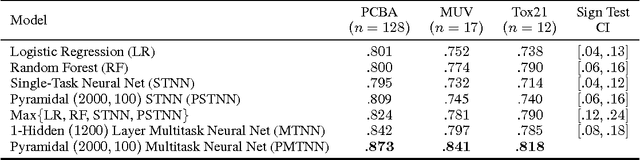
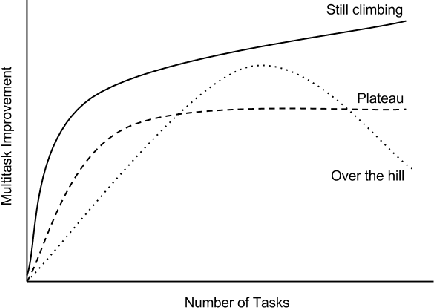
Massively multitask neural architectures provide a learning framework for drug discovery that synthesizes information from many distinct biological sources. To train these architectures at scale, we gather large amounts of data from public sources to create a dataset of nearly 40 million measurements across more than 200 biological targets. We investigate several aspects of the multitask framework by performing a series of empirical studies and obtain some interesting results: (1) massively multitask networks obtain predictive accuracies significantly better than single-task methods, (2) the predictive power of multitask networks improves as additional tasks and data are added, (3) the total amount of data and the total number of tasks both contribute significantly to multitask improvement, and (4) multitask networks afford limited transferability to tasks not in the training set. Our results underscore the need for greater data sharing and further algorithmic innovation to accelerate the drug discovery process.