 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePre-training Graph Neural Networks
May 29, 2019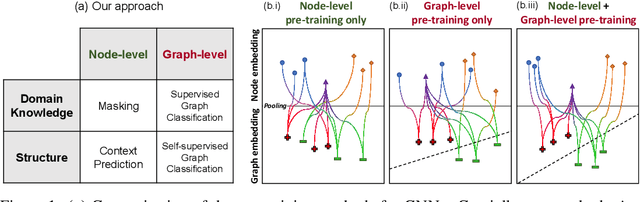
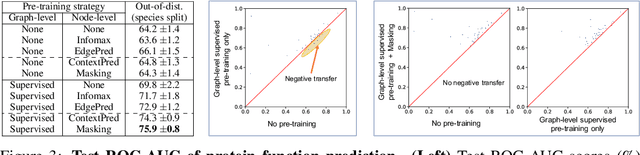
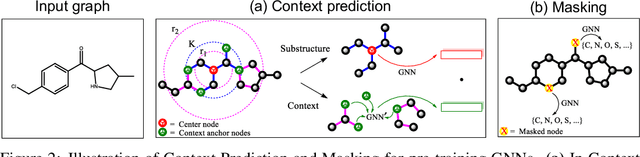
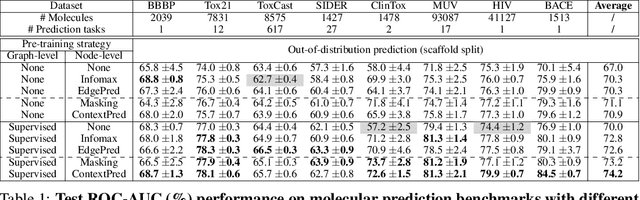
Many applications of machine learning in science and medicine, including molecular property and protein function prediction, can be cast as problems of predicting some properties of graphs, where having good graph representations is critical. However, two key challenges in these domains are (1) extreme scarcity of labeled data due to expensive lab experiments, and (2) needing to extrapolate to test graphs that are structurally different from those seen during training. In this paper, we explore pre-training to address both of these challenges. In particular, working with Graph Neural Networks (GNNs) for representation learning of graphs, we wish to obtain node representations that (1) capture similarity of nodes' network neighborhood structure, (2) can be composed to give accurate graph-level representations, and (3) capture domain-knowledge. To achieve these goals, we propose a series of methods to pre-train GNNs at both the node-level and the graph-level, using both unlabeled data and labeled data from related auxiliary supervised tasks. We perform extensive evaluation on two applications, molecular property and protein function prediction. We observe that performing only graph-level supervised pre-training often leads to marginal performance gain or even can worsen the performance compared to non-pre-trained models. On the other hand, effectively combining both node- and graph-level pre-training techniques significantly improves generalization to out-of-distribution graphs, consistently outperforming non-pre-trained GNNs across 8 datasets in molecular property prediction (resp. 40 tasks in protein function prediction), with the average ROC-AUC improvement of 7.2% (resp. 11.7%).
MoleculeNet: A Benchmark for Molecular Machine Learning
Oct 26, 2018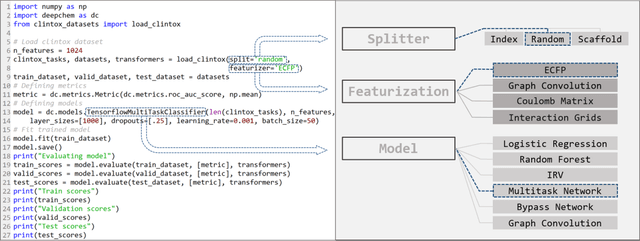
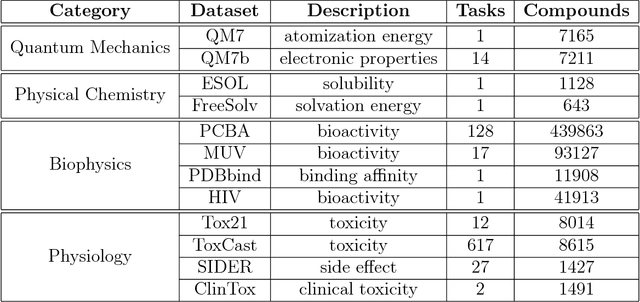
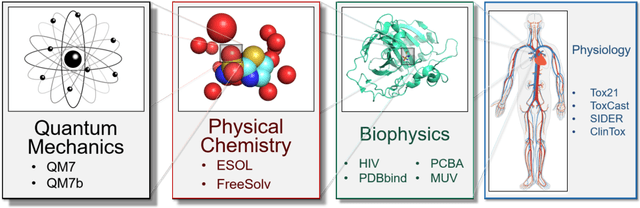
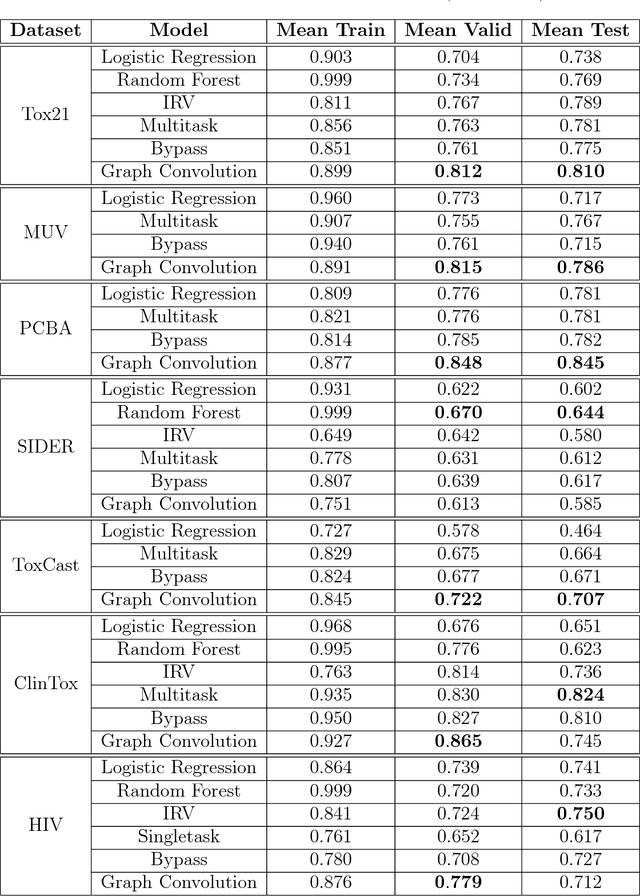
Molecular machine learning has been maturing rapidly over the last few years. Improved methods and the presence of larger datasets have enabled machine learning algorithms to make increasingly accurate predictions about molecular properties. However, algorithmic progress has been limited due to the lack of a standard benchmark to compare the efficacy of proposed methods; most new algorithms are benchmarked on different datasets making it challenging to gauge the quality of proposed methods. This work introduces MoleculeNet, a large scale benchmark for molecular machine learning. MoleculeNet curates multiple public datasets, establishes metrics for evaluation, and offers high quality open-source implementations of multiple previously proposed molecular featurization and learning algorithms (released as part of the DeepChem open source library). MoleculeNet benchmarks demonstrate that learnable representations are powerful tools for molecular machine learning and broadly offer the best performance. However, this result comes with caveats. Learnable representations still struggle to deal with complex tasks under data scarcity and highly imbalanced classification. For quantum mechanical and biophysical datasets, the use of physics-aware featurizations can be more important than choice of particular learning algorithm.
Weakly-Supervised Deep Learning of Heat Transport via Physics Informed Loss
Aug 21, 2018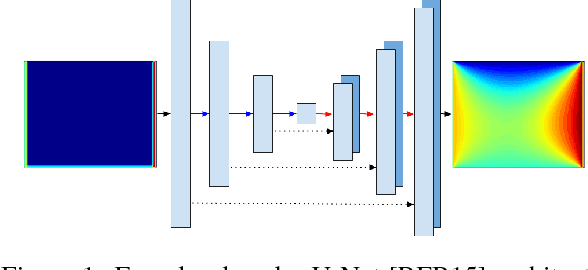
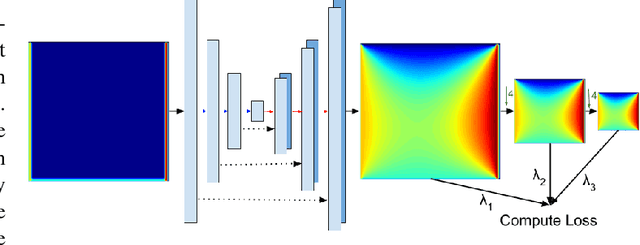
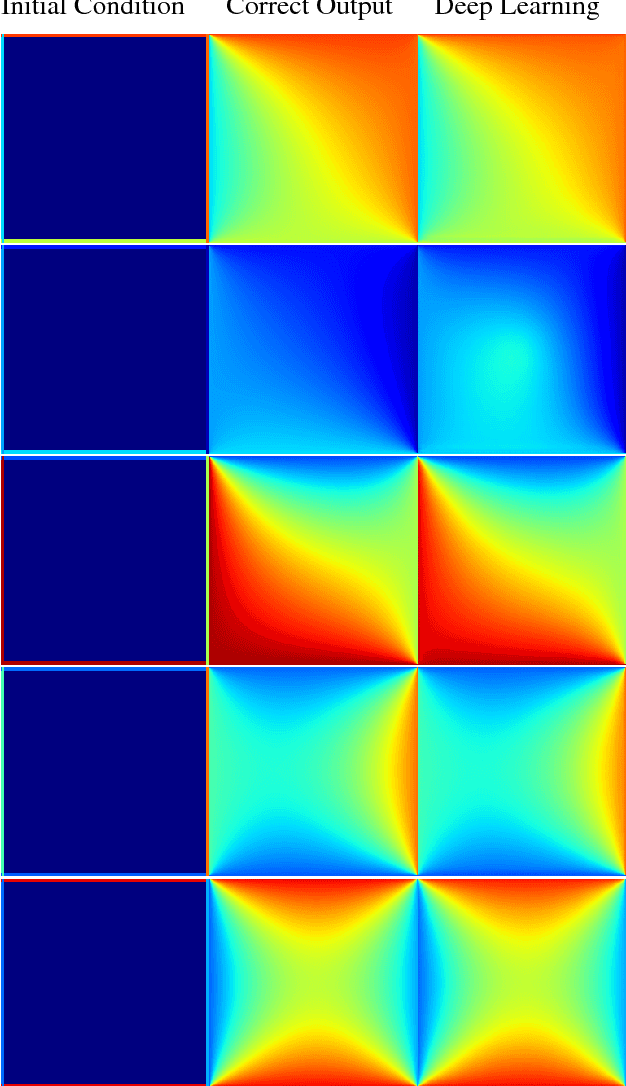
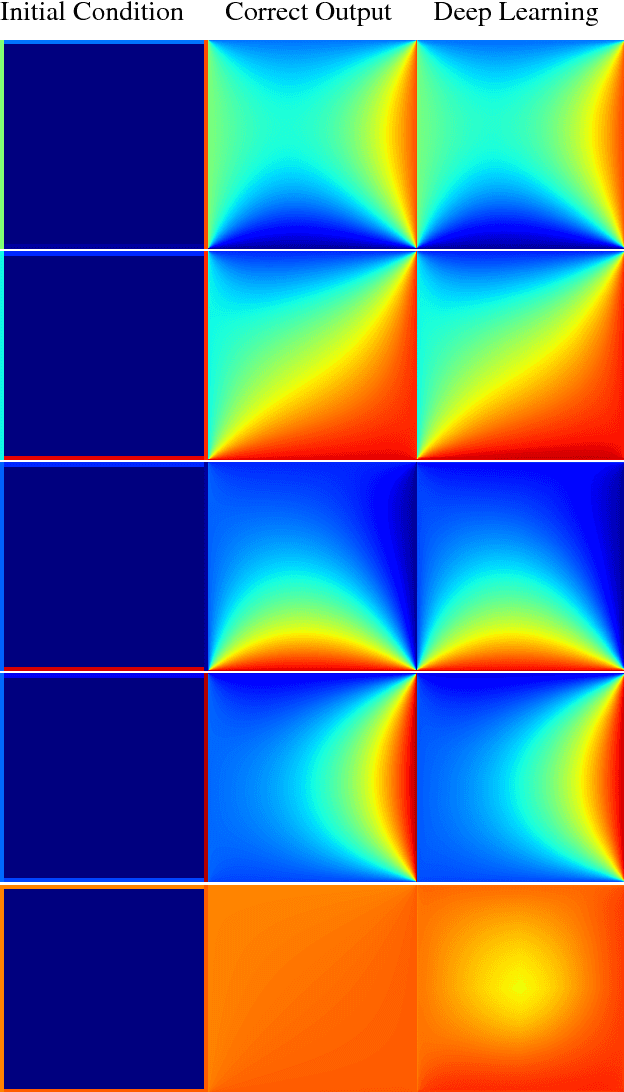
In typical machine learning tasks and applications, it is necessary to obtain or create large labeled datasets in order to to achieve high performance. Unfortunately, large labeled datasets are not always available and can be expensive to source, creating a bottleneck towards more widely applicable machine learning. The paradigm of weak supervision offers an alternative that allows for integration of domain-specific knowledge by enforcing constraints that a correct solution to the learning problem will obey over the output space. In this work, we explore the application of this paradigm to 2-D physical systems governed by non-linear differential equations. We demonstrate that knowledge of the partial differential equations governing a system can be encoded into the loss function of a neural network via an appropriately chosen convolutional kernel. We demonstrate this by showing that the steady-state solution to the 2-D heat equation can be learned directly from initial conditions by a convolutional neural network, in the absence of labeled training data. We also extend recent work in the progressive growing of fully convolutional networks to achieve high accuracy (< 1.5% error) at multiple scales of the heat-flow problem, including at the very large scale (1024x1024). Finally, we demonstrate that this method can be used to speed up exact calculation of the solution to the differential equations via finite difference.
Improved Training with Curriculum GANs
Jul 24, 2018
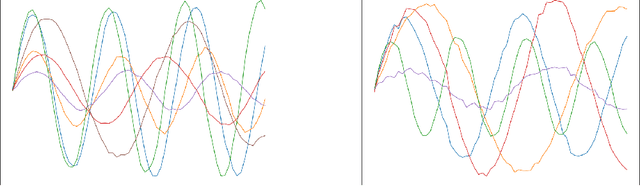
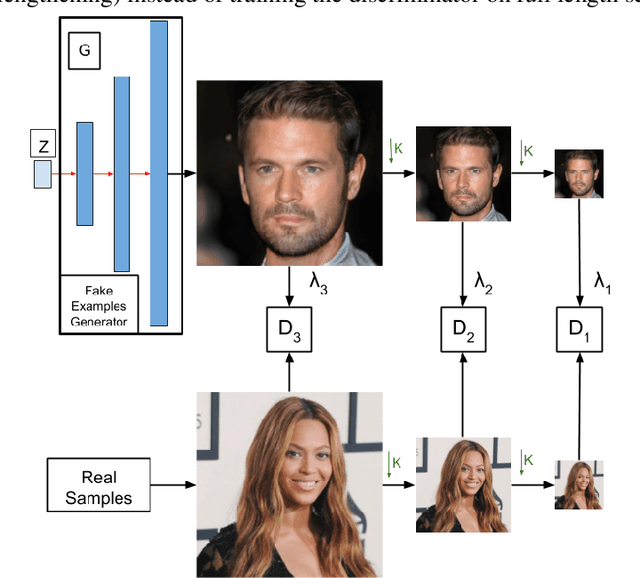

In this paper we introduce Curriculum GANs, a curriculum learning strategy for training Generative Adversarial Networks that increases the strength of the discriminator over the course of training, thereby making the learning task progressively more difficult for the generator. We demonstrate that this strategy is key to obtaining state-of-the-art results in image generation. We also show evidence that this strategy may be broadly applicable to improving GAN training in other data modalities.
Graph Convolutional Policy Network for Goal-Directed Molecular Graph Generation
Jun 07, 2018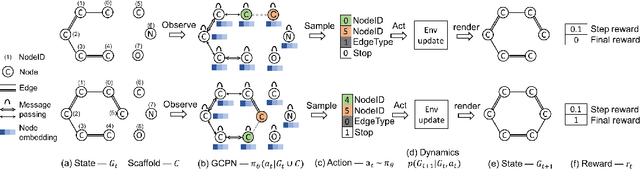
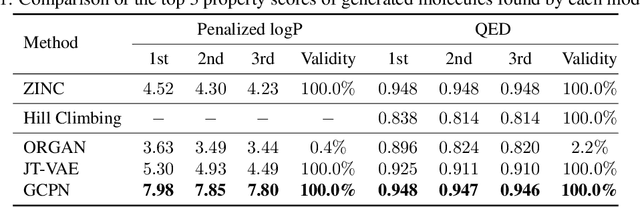
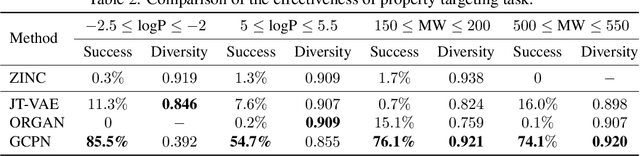
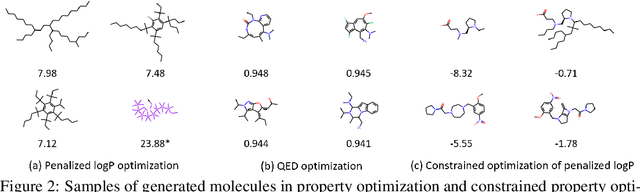
Generating novel graph structures that optimize given objectives while obeying some given underlying rules is fundamental for chemistry, biology and social science research. This is especially important in the task of molecular graph generation, whose goal is to discover novel molecules with desired properties such as drug-likeness and synthetic accessibility, while obeying physical laws such as chemical valency. However, designing models to find molecules that optimize desired properties while incorporating highly complex and non-differentiable rules remains to be a challenging task. Here we propose Graph Convolutional Policy Network (GCPN), a general graph convolutional network based model for goal-directed graph generation through reinforcement learning. The model is trained to optimize domain-specific rewards and adversarial loss through policy gradient, and acts in an environment that incorporates domain-specific rules. Experimental results show that GCPN can achieve 61% improvement on chemical property optimization over state-of-the-art baselines while resembling known molecules, and achieve 184% improvement on the constrained property optimization task.
Retrosynthetic reaction prediction using neural sequence-to-sequence models
Jun 06, 2017

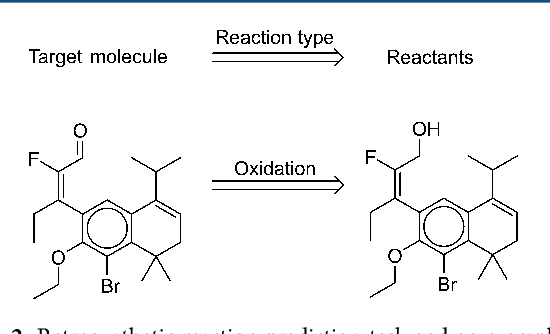
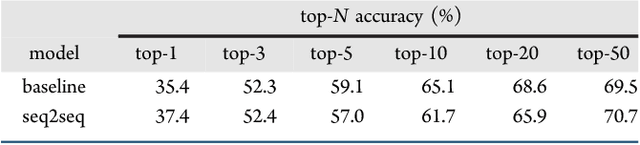
We describe a fully data driven model that learns to perform a retrosynthetic reaction prediction task, which is treated as a sequence-to-sequence mapping problem. The end-to-end trained model has an encoder-decoder architecture that consists of two recurrent neural networks, which has previously shown great success in solving other sequence-to-sequence prediction tasks such as machine translation. The model is trained on 50,000 experimental reaction examples from the United States patent literature, which span 10 broad reaction types that are commonly used by medicinal chemists. We find that our model performs comparably with a rule-based expert system baseline model, and also overcomes certain limitations associated with rule-based expert systems and with any machine learning approach that contains a rule-based expert system component. Our model provides an important first step towards solving the challenging problem of computational retrosynthetic analysis.
Modeling Industrial ADMET Data with Multitask Networks
Jan 13, 2017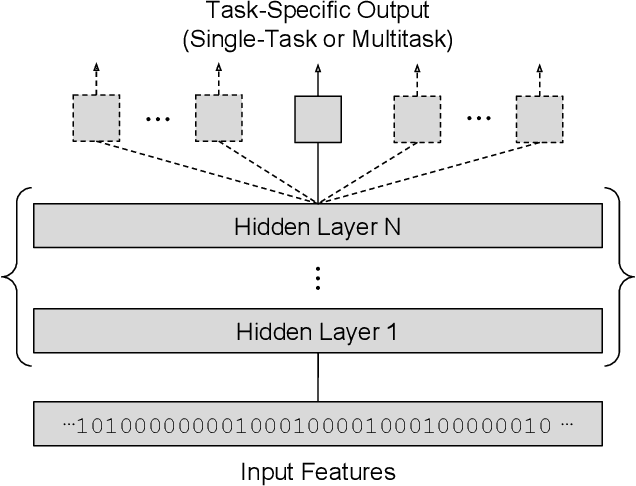
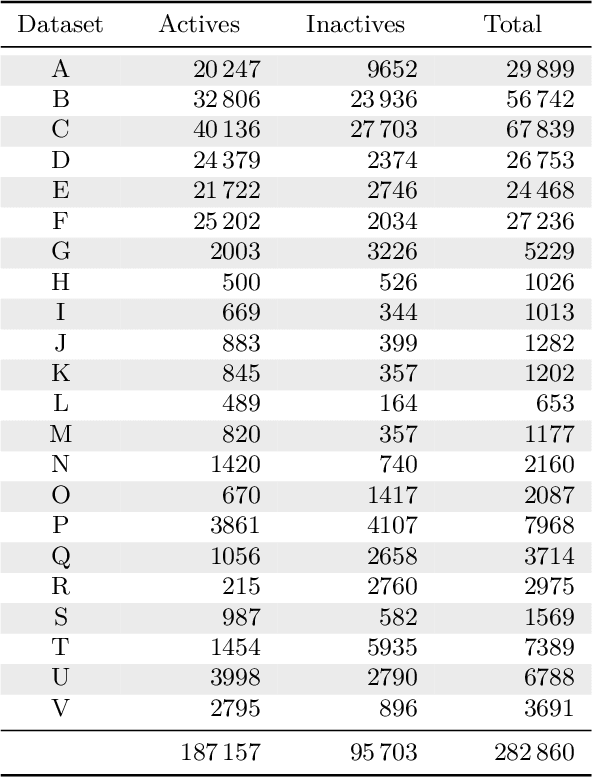
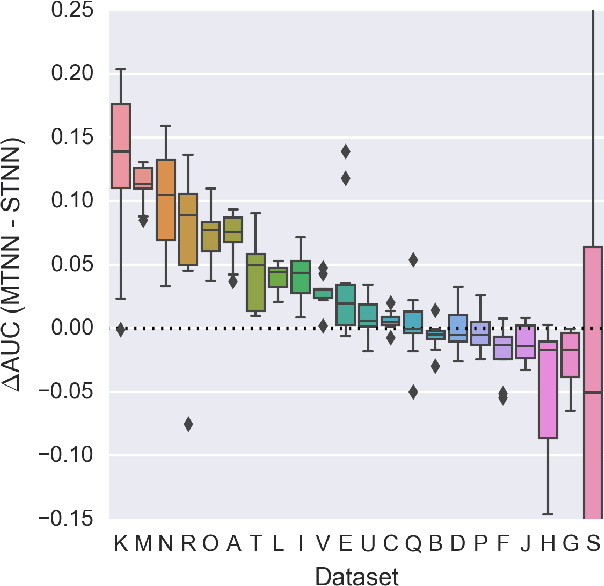
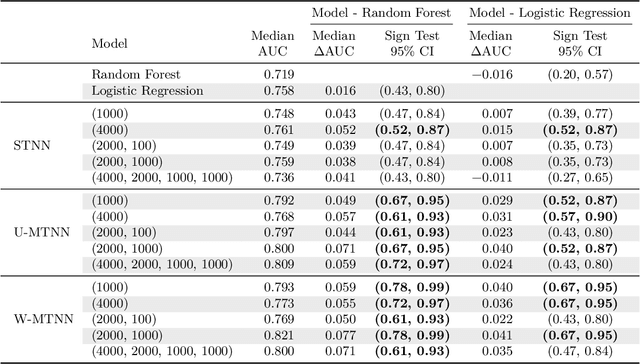
Deep learning methods such as multitask neural networks have recently been applied to ligand-based virtual screening and other drug discovery applications. Using a set of industrial ADMET datasets, we compare neural networks to standard baseline models and analyze multitask learning effects with both random cross-validation and a more relevant temporal validation scheme. We confirm that multitask learning can provide modest benefits over single-task models and show that smaller datasets tend to benefit more than larger datasets from multitask learning. Additionally, we find that adding massive amounts of side information is not guaranteed to improve performance relative to simpler multitask learning. Our results emphasize that multitask effects are highly dataset-dependent, suggesting the use of dataset-specific models to maximize overall performance.
Low Data Drug Discovery with One-shot Learning
Nov 10, 2016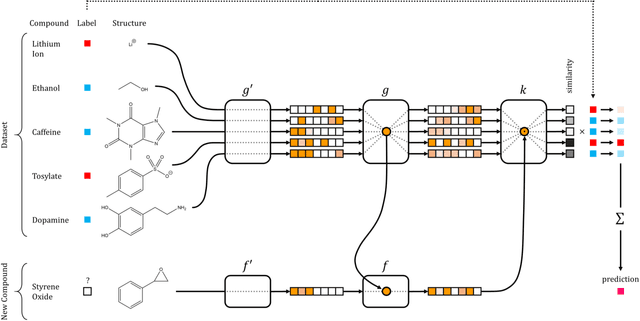
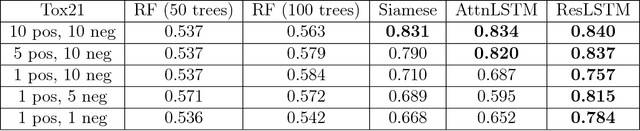
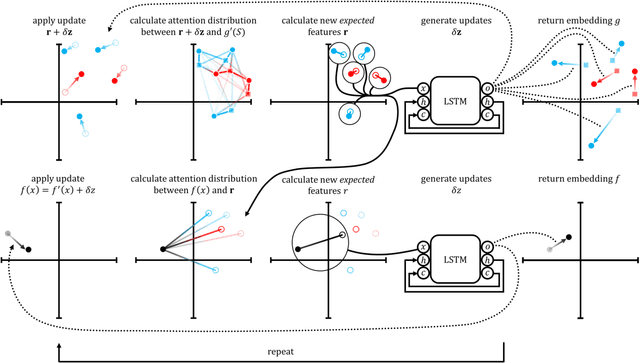
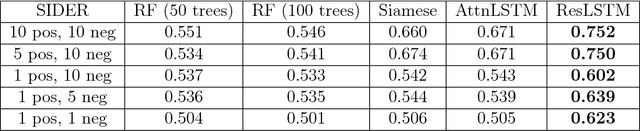
Recent advances in machine learning have made significant contributions to drug discovery. Deep neural networks in particular have been demonstrated to provide significant boosts in predictive power when inferring the properties and activities of small-molecule compounds. However, the applicability of these techniques has been limited by the requirement for large amounts of training data. In this work, we demonstrate how one-shot learning can be used to significantly lower the amounts of data required to make meaningful predictions in drug discovery applications. We introduce a new architecture, the residual LSTM embedding, that, when combined with graph convolutional neural networks, significantly improves the ability to learn meaningful distance metrics over small-molecules. We open source all models introduced in this work as part of DeepChem, an open-source framework for deep-learning in drug discovery.
ROCS-Derived Features for Virtual Screening
Aug 22, 2016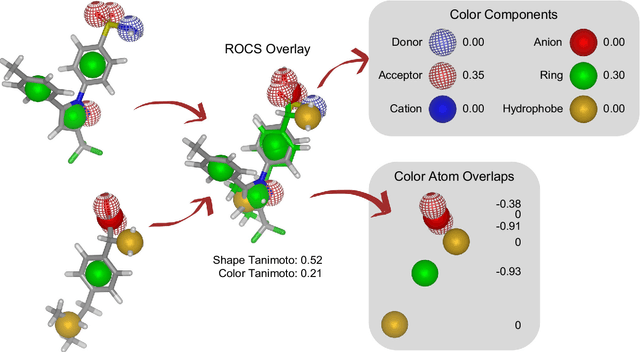

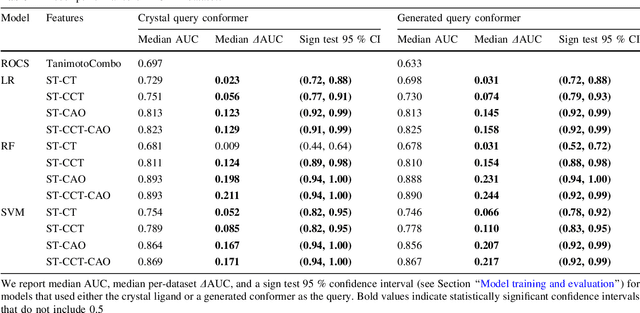
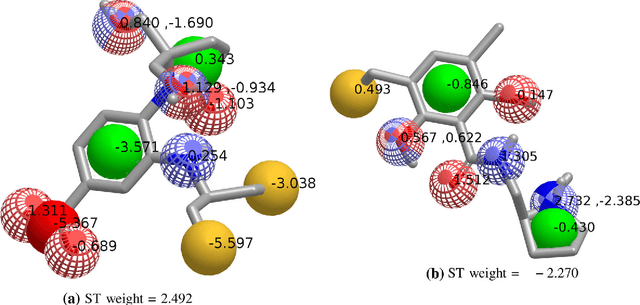
Rapid overlay of chemical structures (ROCS) is a standard tool for the calculation of 3D shape and chemical ("color") similarity. ROCS uses unweighted sums to combine many aspects of similarity, yielding parameter-free models for virtual screening. In this report, we decompose the ROCS color force field into "color components" and "color atom overlaps", novel color similarity features that can be weighted in a system-specific manner by machine learning algorithms. In cross-validation experiments, these additional features significantly improve virtual screening performance (ROC AUC scores) relative to standard ROCS.
Molecular Graph Convolutions: Moving Beyond Fingerprints
Aug 18, 2016
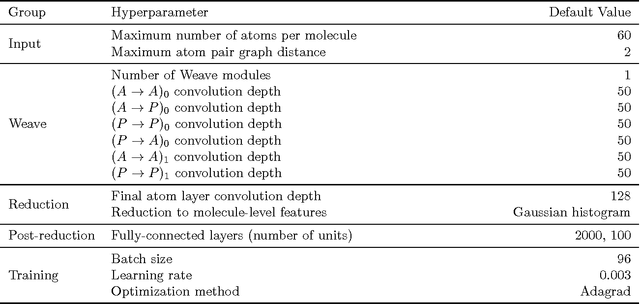
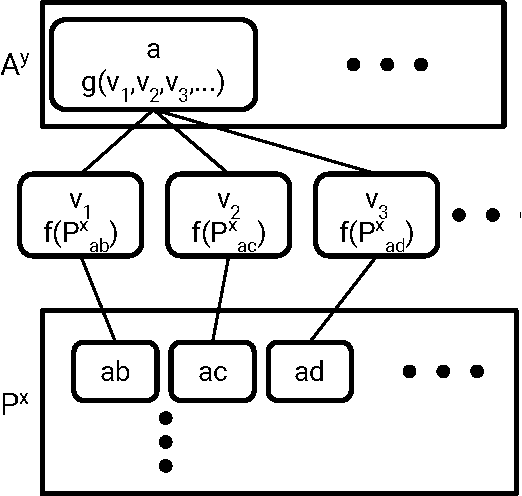
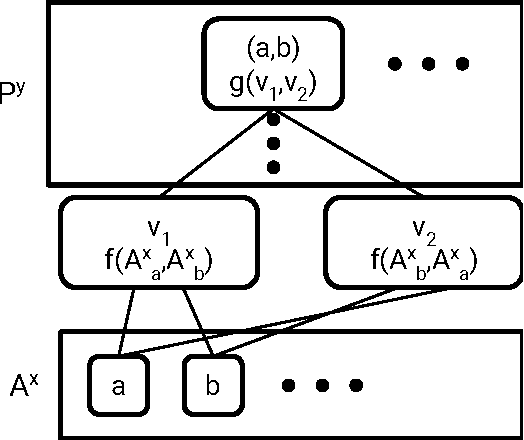
Molecular "fingerprints" encoding structural information are the workhorse of cheminformatics and machine learning in drug discovery applications. However, fingerprint representations necessarily emphasize particular aspects of the molecular structure while ignoring others, rather than allowing the model to make data-driven decisions. We describe molecular "graph convolutions", a machine learning architecture for learning from undirected graphs, specifically small molecules. Graph convolutions use a simple encoding of the molecular graph---atoms, bonds, distances, etc.---which allows the model to take greater advantage of information in the graph structure. Although graph convolutions do not outperform all fingerprint-based methods, they (along with other graph-based methods) represent a new paradigm in ligand-based virtual screening with exciting opportunities for future improvement.
* See "Version information" section