 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient Transition State Searches by Freezing String Method with Graph Neural Network Potentials
Jan 10, 2025Transition states are a critical bottleneck in chemical transformations. Significant efforts have been made to develop algorithms that efficiently locate transition states on potential energy surfaces. However, the computational cost of ab-initio potential energy surface evaluation limits the size of chemical systems that can routinely studied. In this work, we develop and fine-tune a graph neural network potential energy function suitable for describing organic chemical reactions and use it to rapidly identify transition state guess structures. We successfully refine guess structures and locate a transition state in each test system considered and reduce the average number of ab-initio calculations by 47% though use of the graph neural network potential energy function. Our results show that modern machine learning models have reached levels of reliability whereby they can be used to accelerate routine computational chemistry tasks.
Pre-training Graph Neural Networks
May 29, 2019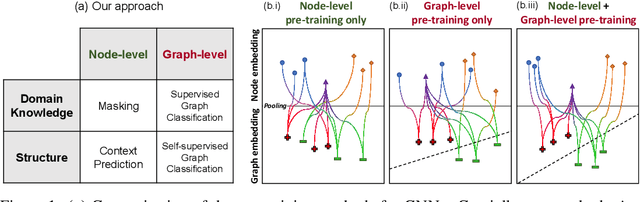
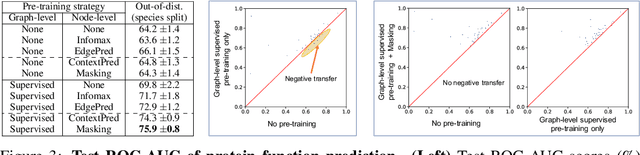
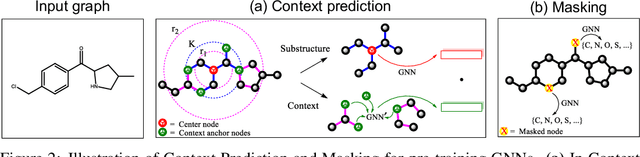
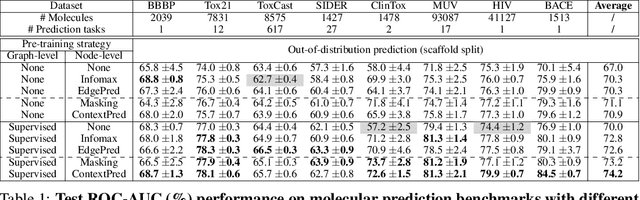
Many applications of machine learning in science and medicine, including molecular property and protein function prediction, can be cast as problems of predicting some properties of graphs, where having good graph representations is critical. However, two key challenges in these domains are (1) extreme scarcity of labeled data due to expensive lab experiments, and (2) needing to extrapolate to test graphs that are structurally different from those seen during training. In this paper, we explore pre-training to address both of these challenges. In particular, working with Graph Neural Networks (GNNs) for representation learning of graphs, we wish to obtain node representations that (1) capture similarity of nodes' network neighborhood structure, (2) can be composed to give accurate graph-level representations, and (3) capture domain-knowledge. To achieve these goals, we propose a series of methods to pre-train GNNs at both the node-level and the graph-level, using both unlabeled data and labeled data from related auxiliary supervised tasks. We perform extensive evaluation on two applications, molecular property and protein function prediction. We observe that performing only graph-level supervised pre-training often leads to marginal performance gain or even can worsen the performance compared to non-pre-trained models. On the other hand, effectively combining both node- and graph-level pre-training techniques significantly improves generalization to out-of-distribution graphs, consistently outperforming non-pre-trained GNNs across 8 datasets in molecular property prediction (resp. 40 tasks in protein function prediction), with the average ROC-AUC improvement of 7.2% (resp. 11.7%).
MoleculeNet: A Benchmark for Molecular Machine Learning
Oct 26, 2018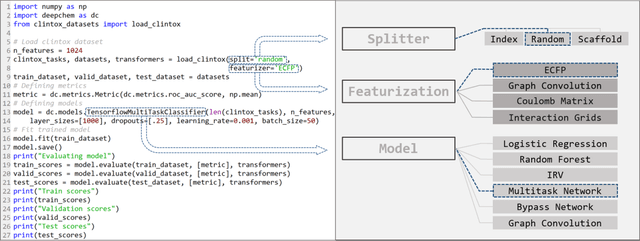
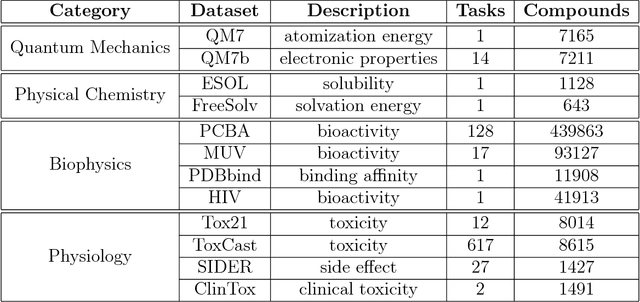
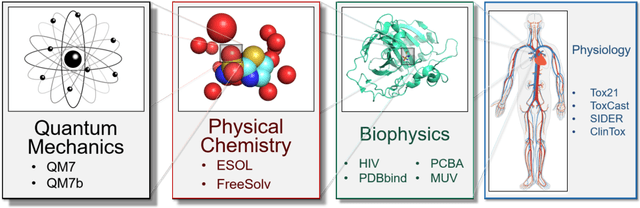
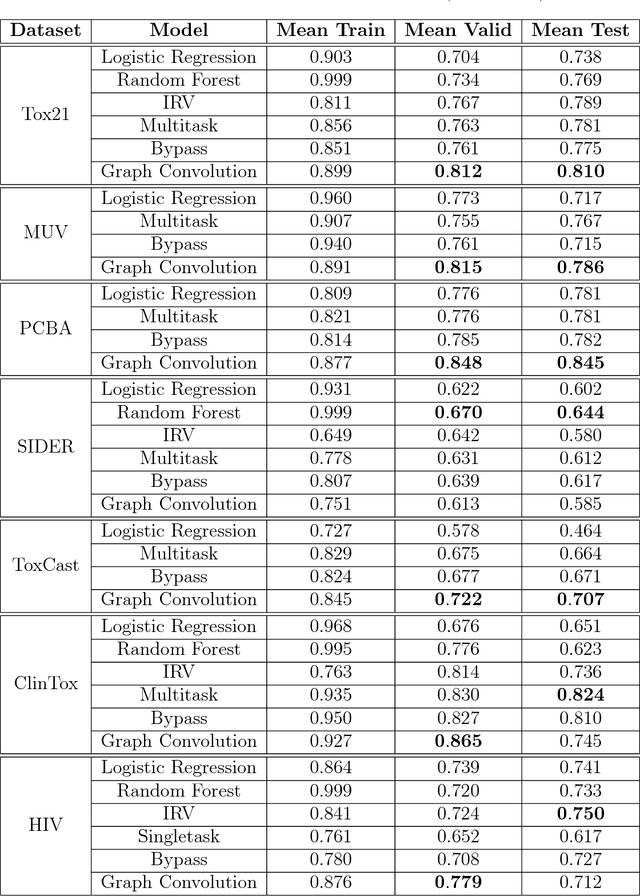
Molecular machine learning has been maturing rapidly over the last few years. Improved methods and the presence of larger datasets have enabled machine learning algorithms to make increasingly accurate predictions about molecular properties. However, algorithmic progress has been limited due to the lack of a standard benchmark to compare the efficacy of proposed methods; most new algorithms are benchmarked on different datasets making it challenging to gauge the quality of proposed methods. This work introduces MoleculeNet, a large scale benchmark for molecular machine learning. MoleculeNet curates multiple public datasets, establishes metrics for evaluation, and offers high quality open-source implementations of multiple previously proposed molecular featurization and learning algorithms (released as part of the DeepChem open source library). MoleculeNet benchmarks demonstrate that learnable representations are powerful tools for molecular machine learning and broadly offer the best performance. However, this result comes with caveats. Learnable representations still struggle to deal with complex tasks under data scarcity and highly imbalanced classification. For quantum mechanical and biophysical datasets, the use of physics-aware featurizations can be more important than choice of particular learning algorithm.
Deep Learning Phase Segregation
Mar 23, 2018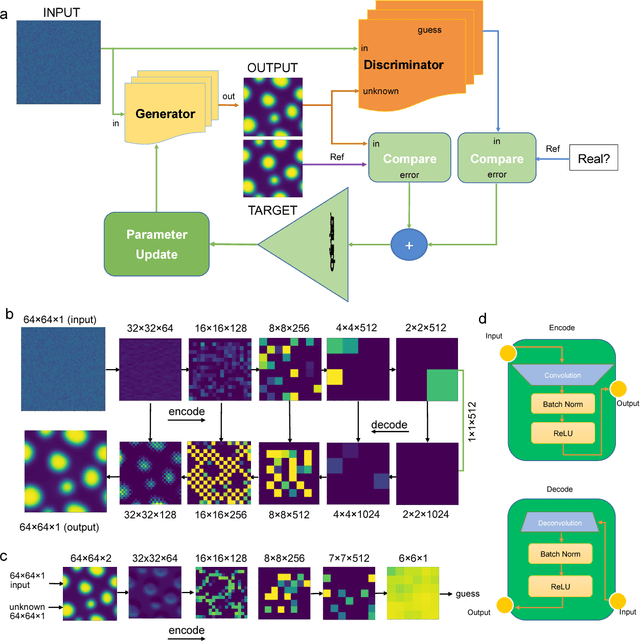
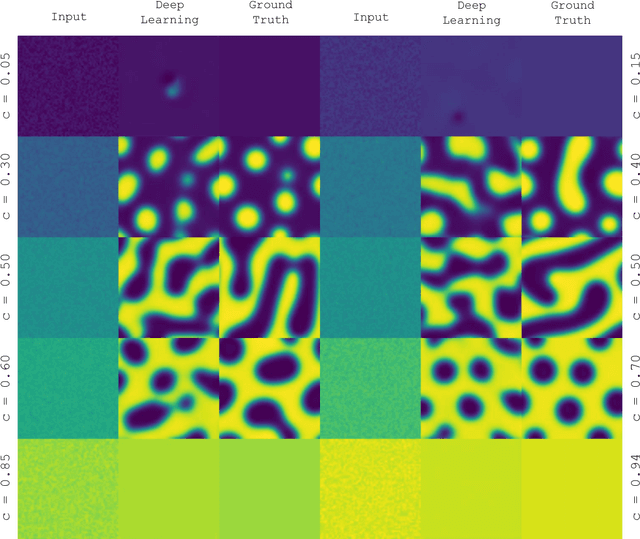


Phase segregation, the process by which the components of a binary mixture spontaneously separate, is a key process in the evolution and design of many chemical, mechanical, and biological systems. In this work, we present a data-driven approach for the learning, modeling, and prediction of phase segregation. A direct mapping between an initially dispersed, immiscible binary fluid and the equilibrium concentration field is learned by conditional generative convolutional neural networks. Concentration field predictions by the deep learning model conserve phase fraction, correctly predict phase transition, and reproduce area, perimeter, and total free energy distributions up to 98% accuracy.
Deep Learning the Physics of Transport Phenomena
Sep 07, 2017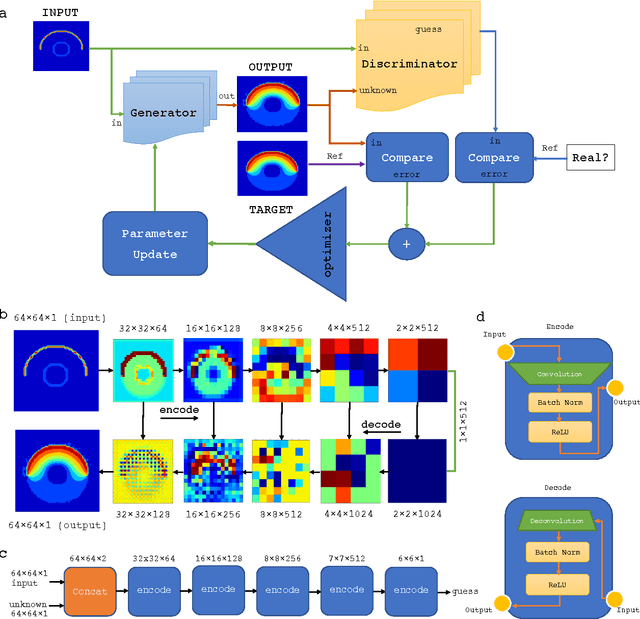
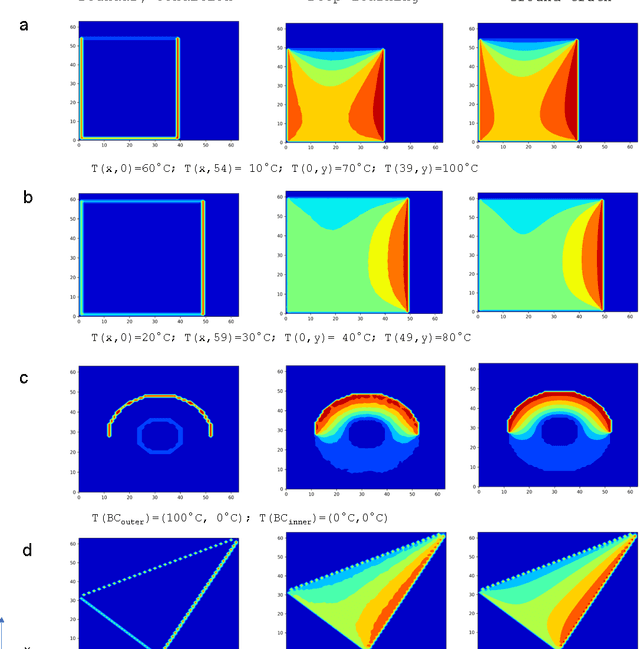
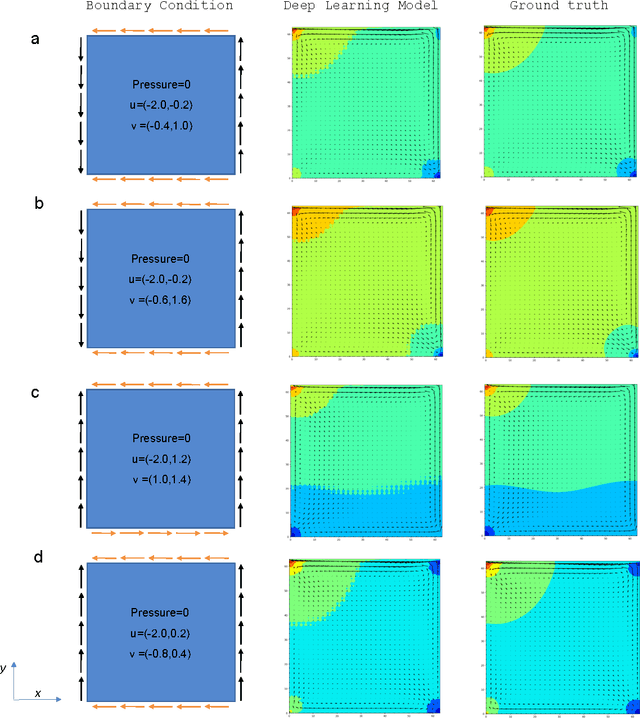
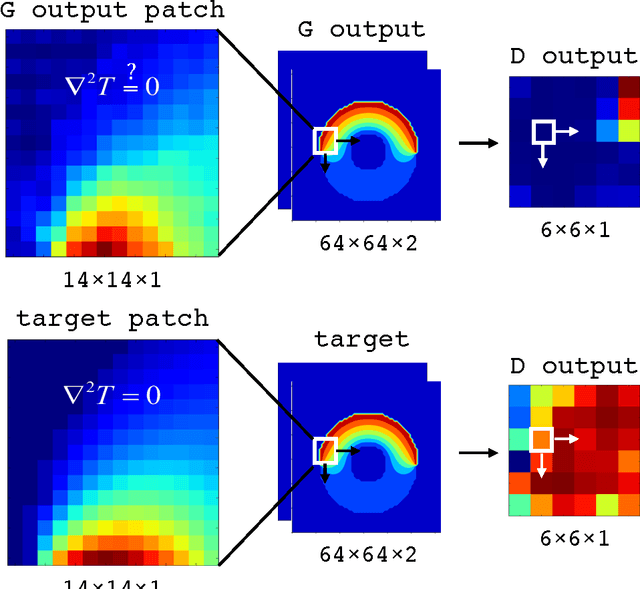
We have developed a new data-driven paradigm for the rapid inference, modeling and simulation of the physics of transport phenomena by deep learning. Using conditional generative adversarial networks (cGAN), we train models for the direct generation of solutions to steady state heat conduction and incompressible fluid flow purely on observation without knowledge of the underlying governing equations. Rather than using iterative numerical methods to approximate the solution of the constitutive equations, cGANs learn to directly generate the solutions to these phenomena, given arbitrary boundary conditions and domain, with high test accuracy (MAE$<$1\%) and state-of-the-art computational performance. The cGAN framework can be used to learn causal models directly from experimental observations where the underlying physical model is complex or unknown.
Retrosynthetic reaction prediction using neural sequence-to-sequence models
Jun 06, 2017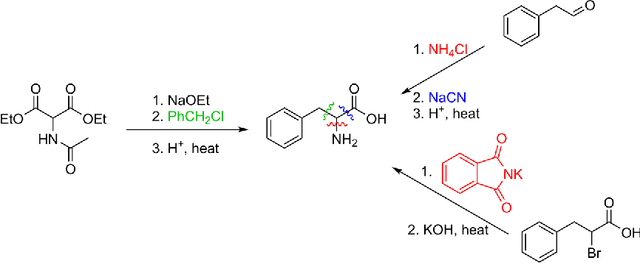

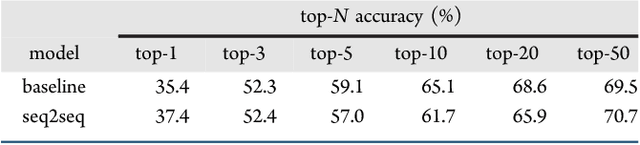
We describe a fully data driven model that learns to perform a retrosynthetic reaction prediction task, which is treated as a sequence-to-sequence mapping problem. The end-to-end trained model has an encoder-decoder architecture that consists of two recurrent neural networks, which has previously shown great success in solving other sequence-to-sequence prediction tasks such as machine translation. The model is trained on 50,000 experimental reaction examples from the United States patent literature, which span 10 broad reaction types that are commonly used by medicinal chemists. We find that our model performs comparably with a rule-based expert system baseline model, and also overcomes certain limitations associated with rule-based expert systems and with any machine learning approach that contains a rule-based expert system component. Our model provides an important first step towards solving the challenging problem of computational retrosynthetic analysis.
Atomic Convolutional Networks for Predicting Protein-Ligand Binding Affinity
Mar 30, 2017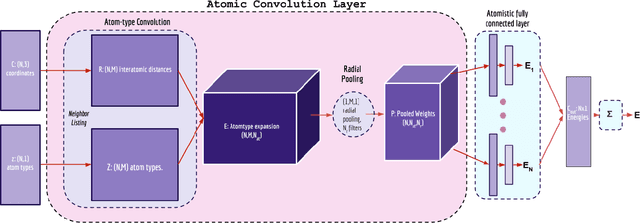

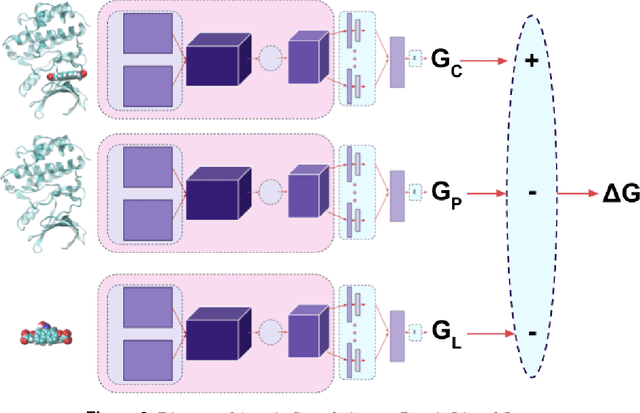

Empirical scoring functions based on either molecular force fields or cheminformatics descriptors are widely used, in conjunction with molecular docking, during the early stages of drug discovery to predict potency and binding affinity of a drug-like molecule to a given target. These models require expert-level knowledge of physical chemistry and biology to be encoded as hand-tuned parameters or features rather than allowing the underlying model to select features in a data-driven procedure. Here, we develop a general 3-dimensional spatial convolution operation for learning atomic-level chemical interactions directly from atomic coordinates and demonstrate its application to structure-based bioactivity prediction. The atomic convolutional neural network is trained to predict the experimentally determined binding affinity of a protein-ligand complex by direct calculation of the energy associated with the complex, protein, and ligand given the crystal structure of the binding pose. Non-covalent interactions present in the complex that are absent in the protein-ligand sub-structures are identified and the model learns the interaction strength associated with these features. We test our model by predicting the binding free energy of a subset of protein-ligand complexes found in the PDBBind dataset and compare with state-of-the-art cheminformatics and machine learning-based approaches. We find that all methods achieve experimental accuracy and that atomic convolutional networks either outperform or perform competitively with the cheminformatics based methods. Unlike all previous protein-ligand prediction systems, atomic convolutional networks are end-to-end and fully-differentiable. They represent a new data-driven, physics-based deep learning model paradigm that offers a strong foundation for future improvements in structure-based bioactivity prediction.