 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMoleculeNet: A Benchmark for Molecular Machine Learning
Oct 26, 2018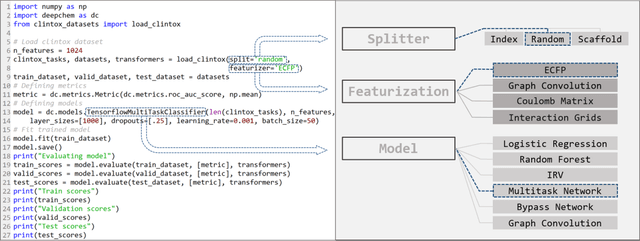
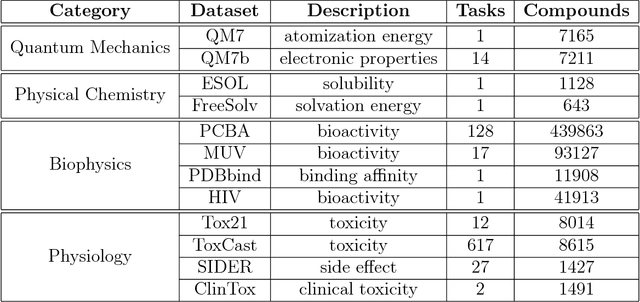
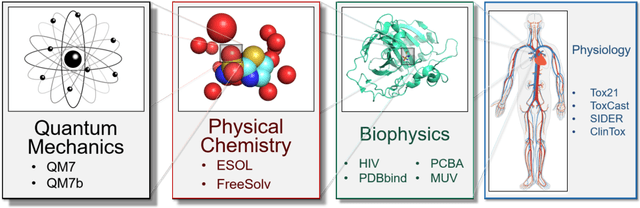
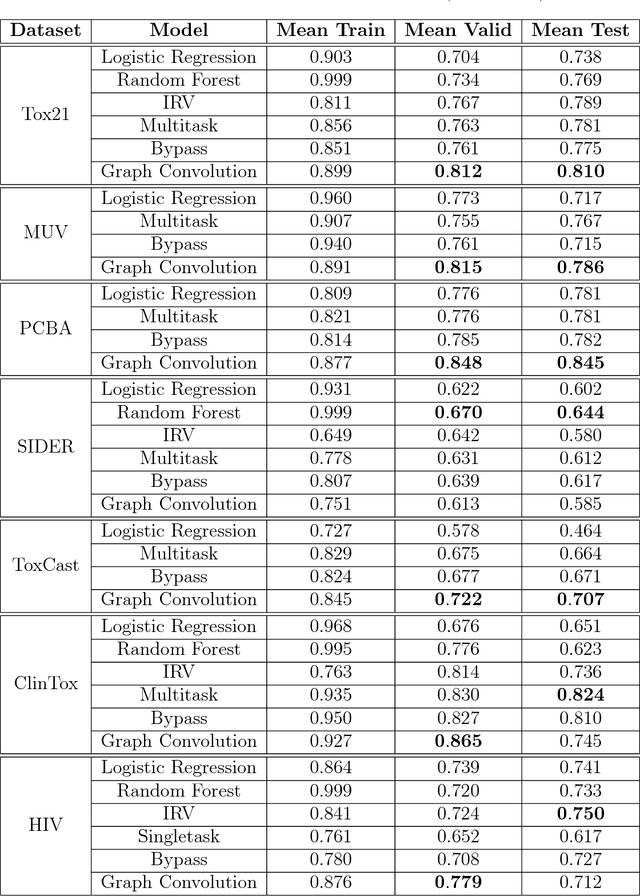
Molecular machine learning has been maturing rapidly over the last few years. Improved methods and the presence of larger datasets have enabled machine learning algorithms to make increasingly accurate predictions about molecular properties. However, algorithmic progress has been limited due to the lack of a standard benchmark to compare the efficacy of proposed methods; most new algorithms are benchmarked on different datasets making it challenging to gauge the quality of proposed methods. This work introduces MoleculeNet, a large scale benchmark for molecular machine learning. MoleculeNet curates multiple public datasets, establishes metrics for evaluation, and offers high quality open-source implementations of multiple previously proposed molecular featurization and learning algorithms (released as part of the DeepChem open source library). MoleculeNet benchmarks demonstrate that learnable representations are powerful tools for molecular machine learning and broadly offer the best performance. However, this result comes with caveats. Learnable representations still struggle to deal with complex tasks under data scarcity and highly imbalanced classification. For quantum mechanical and biophysical datasets, the use of physics-aware featurizations can be more important than choice of particular learning algorithm.
Low Data Drug Discovery with One-shot Learning
Nov 10, 2016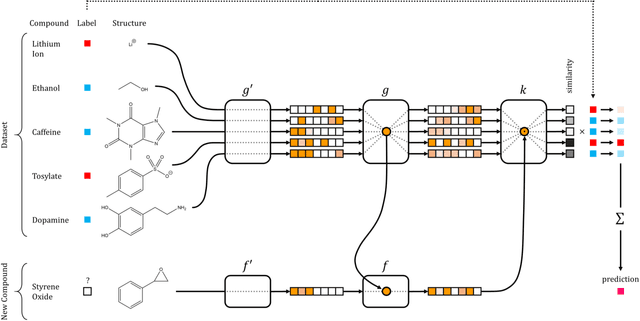
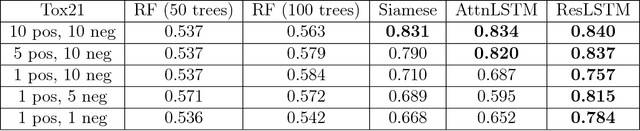
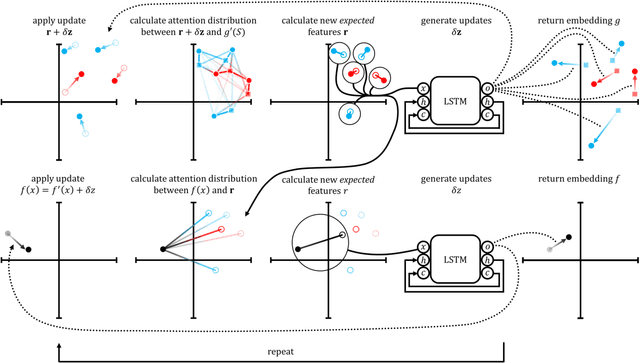
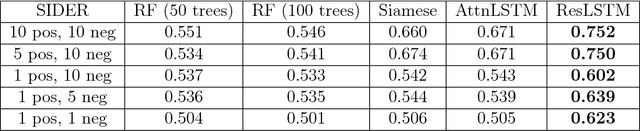
Recent advances in machine learning have made significant contributions to drug discovery. Deep neural networks in particular have been demonstrated to provide significant boosts in predictive power when inferring the properties and activities of small-molecule compounds. However, the applicability of these techniques has been limited by the requirement for large amounts of training data. In this work, we demonstrate how one-shot learning can be used to significantly lower the amounts of data required to make meaningful predictions in drug discovery applications. We introduce a new architecture, the residual LSTM embedding, that, when combined with graph convolutional neural networks, significantly improves the ability to learn meaningful distance metrics over small-molecules. We open source all models introduced in this work as part of DeepChem, an open-source framework for deep-learning in drug discovery.