 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStep Change Improvement in ADMET Prediction with PotentialNet Deep Featurization
Mar 28, 2019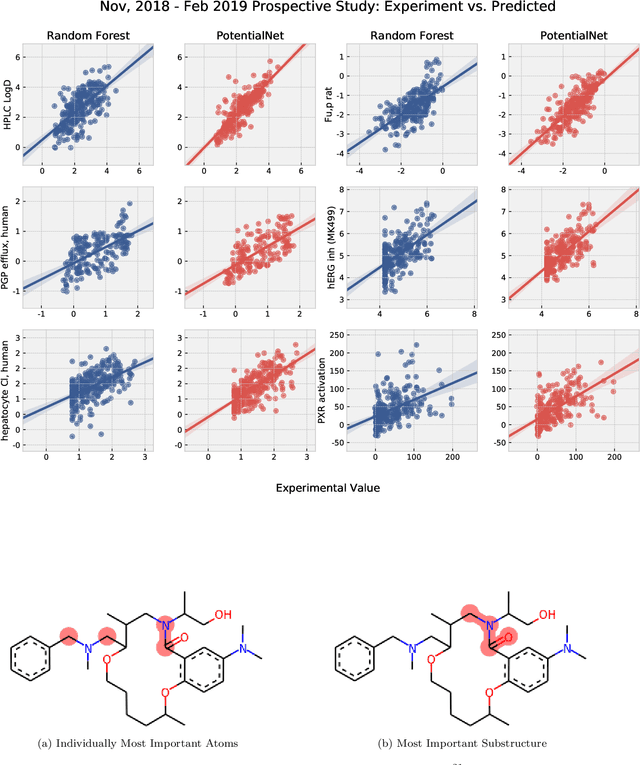
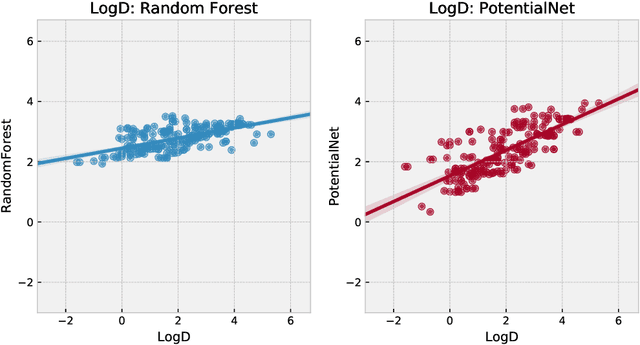
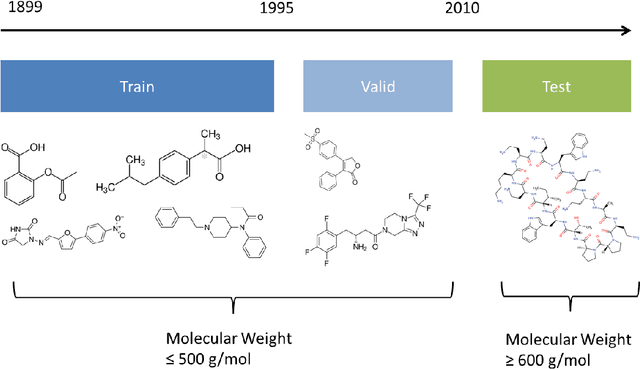
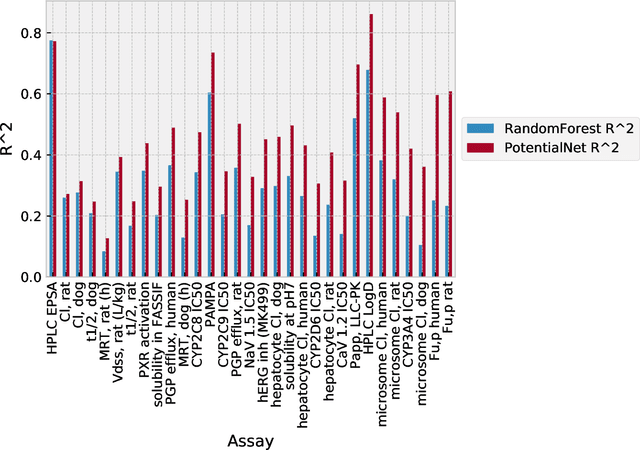
The Absorption, Distribution, Metabolism, Elimination, and Toxicity (ADMET) properties of drug candidates are estimated to account for up to 50% of all clinical trial failures. Predicting ADMET properties has therefore been of great interest to the cheminformatics and medicinal chemistry communities in recent decades. Traditional cheminformatics approaches, whether the learner is a random forest or a deep neural network, leverage fixed fingerprint feature representations of molecules. In contrast, in this paper, we learn the features most relevant to each chemical task at hand by representing each molecule explicitly as a graph, where each node is an atom and each edge is a bond. By applying graph convolutions to this explicit molecular representation, we achieve, to our knowledge, unprecedented accuracy in prediction of ADMET properties. By challenging our methodology with rigorous cross-validation procedures and prospective analyses, we show that deep featurization better enables molecular predictors to not only interpolate but also extrapolate to new regions of chemical space.
MoleculeNet: A Benchmark for Molecular Machine Learning
Oct 26, 2018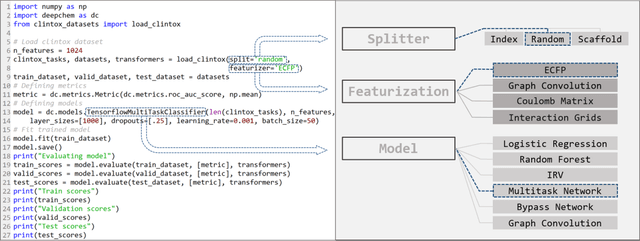
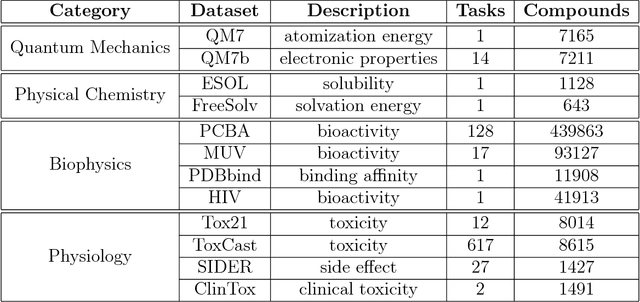
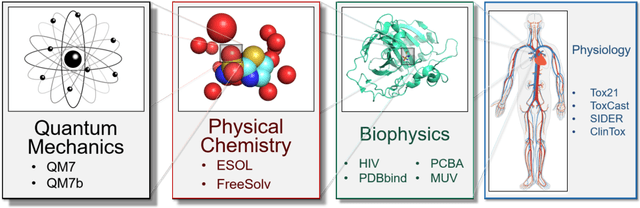
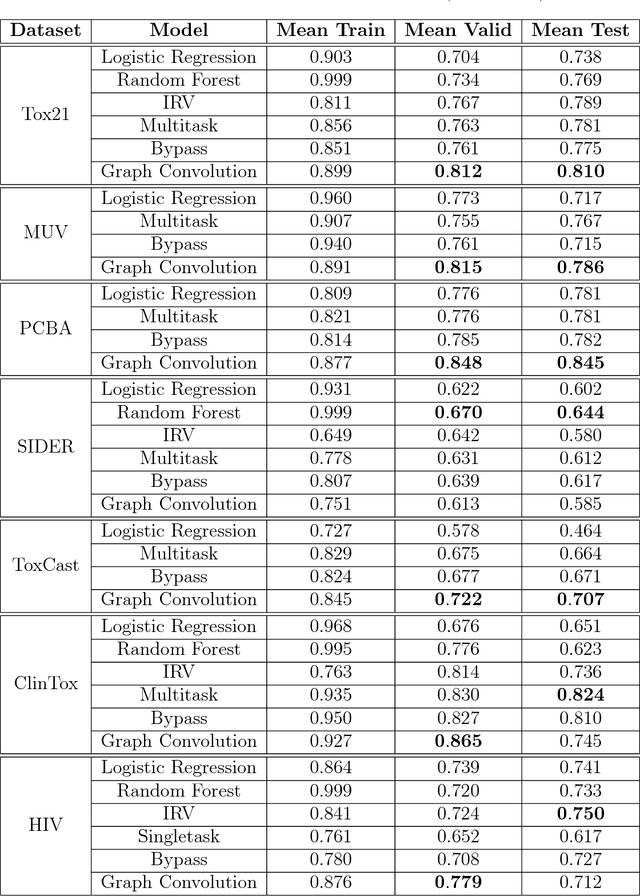
Molecular machine learning has been maturing rapidly over the last few years. Improved methods and the presence of larger datasets have enabled machine learning algorithms to make increasingly accurate predictions about molecular properties. However, algorithmic progress has been limited due to the lack of a standard benchmark to compare the efficacy of proposed methods; most new algorithms are benchmarked on different datasets making it challenging to gauge the quality of proposed methods. This work introduces MoleculeNet, a large scale benchmark for molecular machine learning. MoleculeNet curates multiple public datasets, establishes metrics for evaluation, and offers high quality open-source implementations of multiple previously proposed molecular featurization and learning algorithms (released as part of the DeepChem open source library). MoleculeNet benchmarks demonstrate that learnable representations are powerful tools for molecular machine learning and broadly offer the best performance. However, this result comes with caveats. Learnable representations still struggle to deal with complex tasks under data scarcity and highly imbalanced classification. For quantum mechanical and biophysical datasets, the use of physics-aware featurizations can be more important than choice of particular learning algorithm.
PotentialNet for Molecular Property Prediction
Oct 22, 2018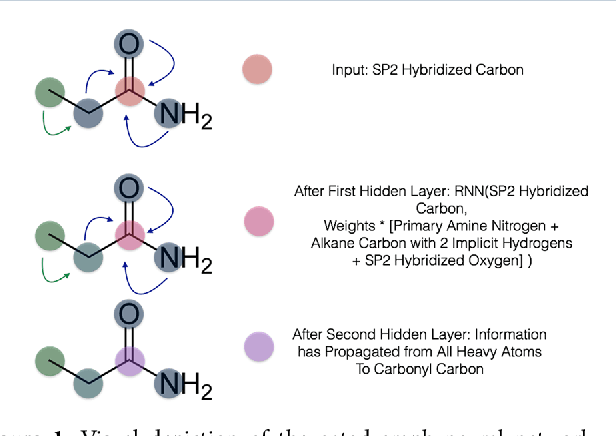
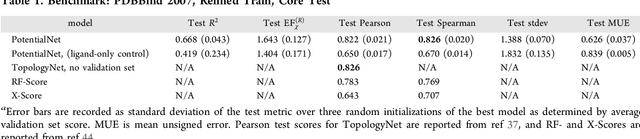
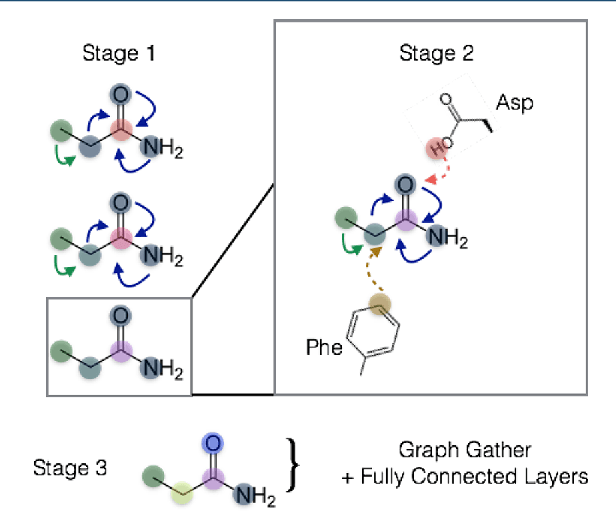
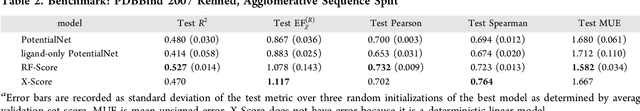
The arc of drug discovery entails a multiparameter optimization problem spanning vast length scales. They key parameters range from solubility (angstroms) to protein-ligand binding (nanometers) to in vivo toxicity (meters). Through feature learning---instead of feature engineering---deep neural networks promise to outperform both traditional physics-based and knowledge-based machine learning models for predicting molecular properties pertinent to drug discovery. To this end, we present the PotentialNet family of graph convolutions. These models are specifically designed for and achieve state-of-the-art performance for protein-ligand binding affinity. We further validate these deep neural networks by setting new standards of performance in several ligand-based tasks. In parallel, we introduce a new metric, the Regression Enrichment Factor $EF_\chi^{(R)}$, to measure the early enrichment of computational models for chemical data. Finally, we introduce a cross-validation strategy based on structural homology clustering that can more accurately measure model generalizability, which crucially distinguishes the aims of machine learning for drug discovery from standard machine learning tasks.
Machine Learning Harnesses Molecular Dynamics to Discover New $μ$ Opioid Chemotypes
Mar 12, 2018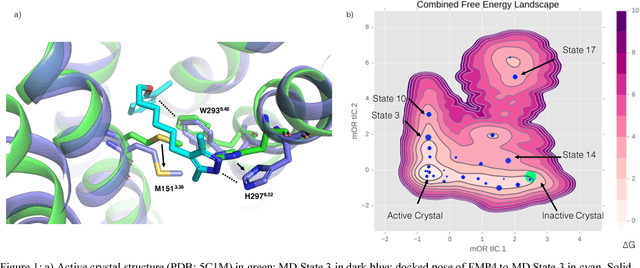
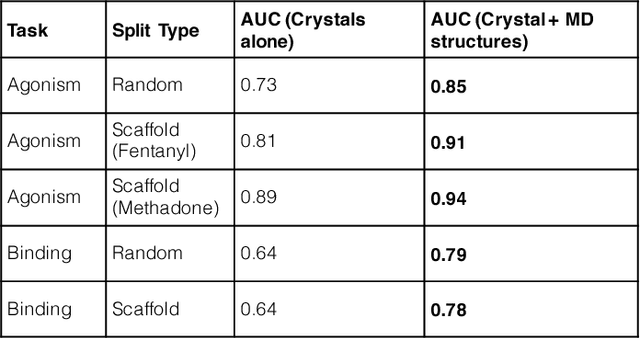
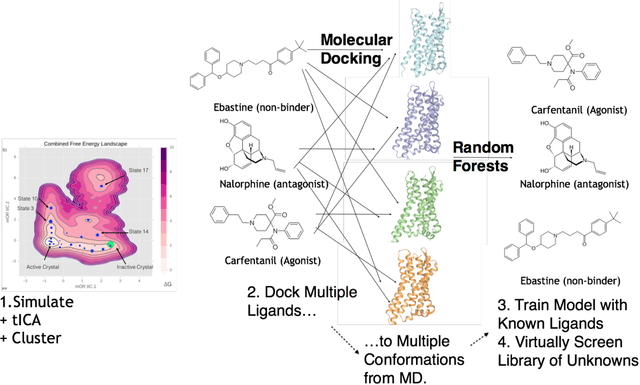
Computational chemists typically assay drug candidates by virtually screening compounds against crystal structures of a protein despite the fact that some targets, like the $\mu$ Opioid Receptor and other members of the GPCR family, traverse many non-crystallographic states. We discover new conformational states of $\mu OR$ with molecular dynamics simulation and then machine learn ligand-structure relationships to predict opioid ligand function. These artificial intelligence models identified a novel $\mu$ opioid chemotype.
Atomic Convolutional Networks for Predicting Protein-Ligand Binding Affinity
Mar 30, 2017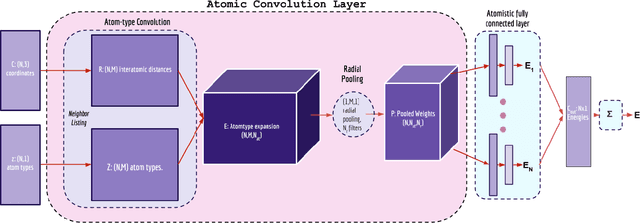

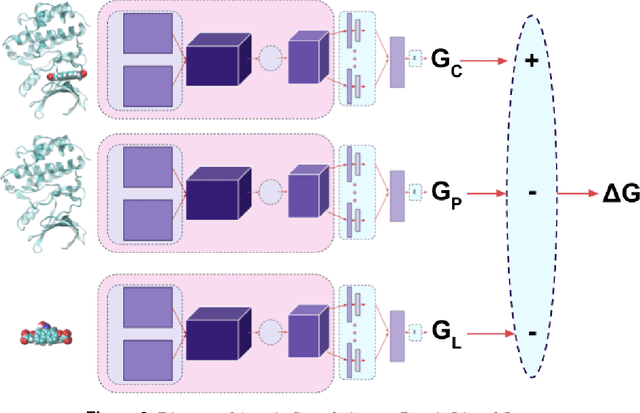

Empirical scoring functions based on either molecular force fields or cheminformatics descriptors are widely used, in conjunction with molecular docking, during the early stages of drug discovery to predict potency and binding affinity of a drug-like molecule to a given target. These models require expert-level knowledge of physical chemistry and biology to be encoded as hand-tuned parameters or features rather than allowing the underlying model to select features in a data-driven procedure. Here, we develop a general 3-dimensional spatial convolution operation for learning atomic-level chemical interactions directly from atomic coordinates and demonstrate its application to structure-based bioactivity prediction. The atomic convolutional neural network is trained to predict the experimentally determined binding affinity of a protein-ligand complex by direct calculation of the energy associated with the complex, protein, and ligand given the crystal structure of the binding pose. Non-covalent interactions present in the complex that are absent in the protein-ligand sub-structures are identified and the model learns the interaction strength associated with these features. We test our model by predicting the binding free energy of a subset of protein-ligand complexes found in the PDBBind dataset and compare with state-of-the-art cheminformatics and machine learning-based approaches. We find that all methods achieve experimental accuracy and that atomic convolutional networks either outperform or perform competitively with the cheminformatics based methods. Unlike all previous protein-ligand prediction systems, atomic convolutional networks are end-to-end and fully-differentiable. They represent a new data-driven, physics-based deep learning model paradigm that offers a strong foundation for future improvements in structure-based bioactivity prediction.